|
Der Weg zur zellspezifischen mRNA: vom Chromomer zum Transkriptosom.
Vorbemerkung
Ein großes Problem der Genetik ist die Definition von Anfang und Ende eines Primärtranskriptes210, da meines Wissens noch niemand ein solches Primärtranskript und den Weg seiner Entstehung in vivo beobachten oder analysieren konnte. Die Festlegung des Transkriptionsstartpunktes (TSS: transcription-start-site) ist deshalb ebenso wie die Bestimmung des Promotors oder des aktiven poly-A-Signals mit großen Schwierigkeiten verbunden. Die mRNA, die man aus einem Gewebe oder einer Zellkultur isoliert, ist das Endprodukt eines Prozesses, von dem man lediglich weiß, dass an seinem Anfang der Initiationskomplex und am Ende die reife mRNA steht was dazwischen geschieht, davon weiß man wenig, wenn man vom Prozeß des Spleißens einmal absieht, dessen basaler Mechanismus ziemlich gut untersucht ist. Auch der cDNA211, die man von der zu untersuchenden mRNA mit Hilfe der Reversen Transkriptase herstellen kann, kommt nur eine sehr geringe Aussagekraft für die Struktur des Primärtranskripts zu.
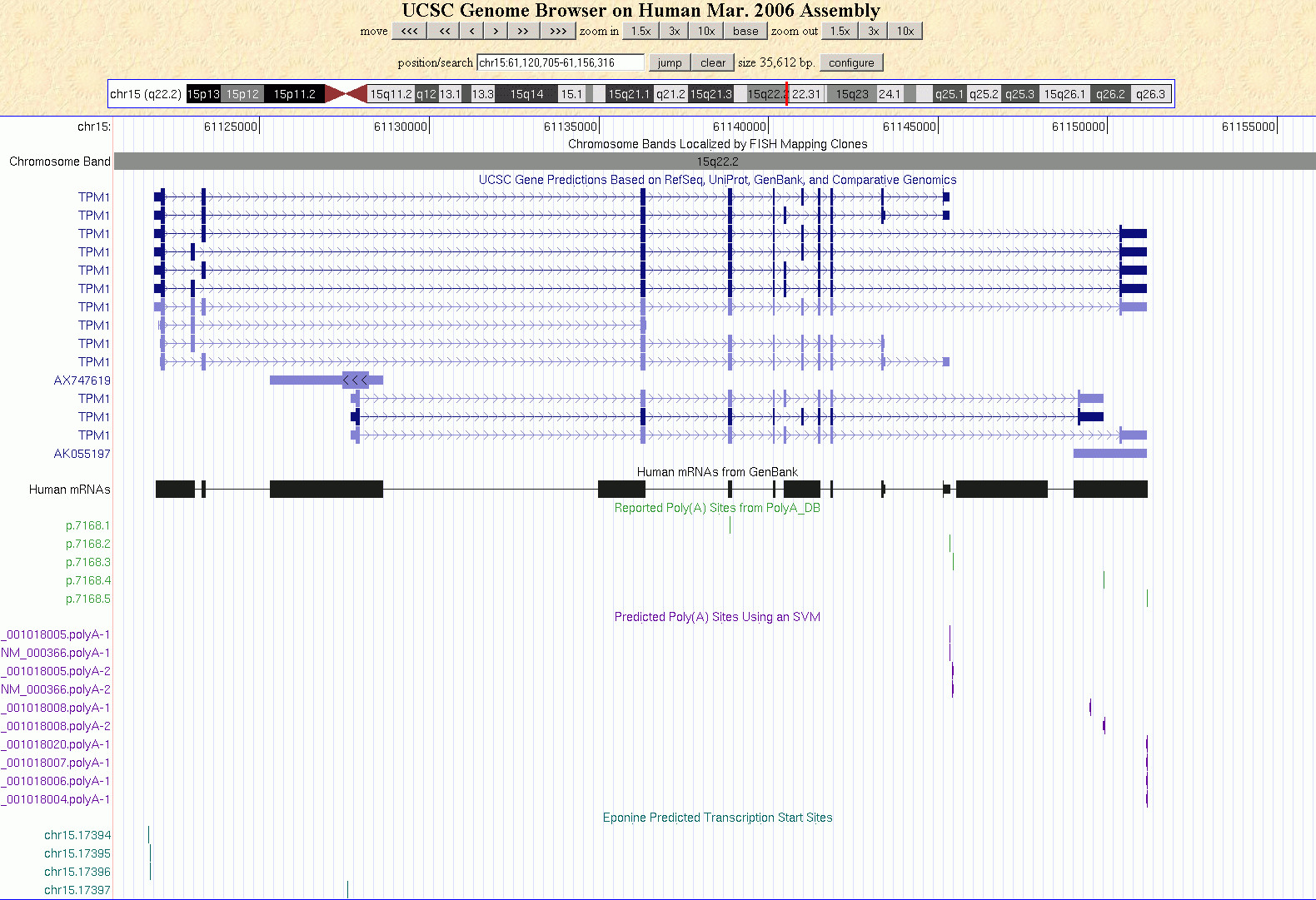
Erst in den letzten Jahren hat sich allmählich die Erkenntnis durchgesetzt, dass Gene in der Regel mehrere TSS haben und es sind viele Versuche gemacht worden, Promotoren und/oder die Transkriptionsstartpunkte von Genen mit Hilfe unterschiedlicher Methoden und Ansätze zu zu identifizieren. Eponine, PromotorInspector, TATA-2.6 und TATA-6.5 sind nur einige der Programme und 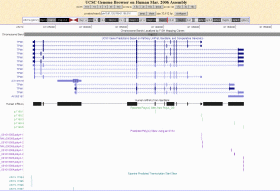 Methoden, die entwickelt wurden, um eine Aussage über die Lage von Promotoren oder die TSS von Genen zu machen. Alle Programme rühmen sich einer mehr oder weniger großen Erkennungsrate, die von ca. 60% bis etwas über 73 % reichen soll. Eines ist allen genannten Methoden gemeinsam: sie entdecken fast ausschließlich putative TSS, die unmittelbar vor dem ersten Exon des jeweiligen Gens liegen. Die Abbildung zeigt das TPM1-Gen im UCSC Genome Browser mit den von Eponine vorhergesagten TSS und poly-Adenylierungsstellen, die zum einen auf Vorhersage der SVM (support vector machine) zum anderen auf Daten der polyA_DB beruhen (http://polya.umdnj.edu/polyadb). Methoden, die entwickelt wurden, um eine Aussage über die Lage von Promotoren oder die TSS von Genen zu machen. Alle Programme rühmen sich einer mehr oder weniger großen Erkennungsrate, die von ca. 60% bis etwas über 73 % reichen soll. Eines ist allen genannten Methoden gemeinsam: sie entdecken fast ausschließlich putative TSS, die unmittelbar vor dem ersten Exon des jeweiligen Gens liegen. Die Abbildung zeigt das TPM1-Gen im UCSC Genome Browser mit den von Eponine vorhergesagten TSS und poly-Adenylierungsstellen, die zum einen auf Vorhersage der SVM (support vector machine) zum anderen auf Daten der polyA_DB beruhen (http://polya.umdnj.edu/polyadb).
Wie ich weiter oben schon einmal gesagt habe, müssen Chromatin-Domänen und Chromatin-Subdomänen das folgt logisch aus der in dieser Arbeit analysierten und beschriebenen Struktur der DNA - die DNA-Abschnitte sein, in welchen alle für die zellspezifische Expression eines Gens entscheidenden Sequenzen durch eine gemeinsame Nukleosomengrösse zu einer Funktionseinheit zusammengefasst sind: die hotspots, die zur Definition einer bestimmten Transkriptionsschleife, zur Transkription einer hnRNA und durch processing dieser hnRNA zu einer bestimmten mRNA führen, sind also wie ich von Anfang an postuliert habe und im nächsten Kapitel auch belegen werde innerhalb einer bestimmten Chromatin-Domäne über eine definierte Nukleosomengrösse logisch miteinander verknüpft.
Auf Grund der Überlegungen und Zusammenhänge, die ich in Teil II und am Anfang dieses Teils III diskutiert habe, können aktivierbare Promotoren und poly-A-Signale eines Gens nur in einem umschriebenen Bereich downstream und upstream des jeweiligen Gens liegen, der durch die Grenzen von Subdomänen-SMARs der Klasse III definiert wird.
Für die Regulation des TPM1-Gens ergibt sich so eine DNA-Sequenz (ohne die SMARS am Beginn und am Ende der Subdomäne) aus 39634 Basenpaaren, die beim Basenpaar 61113922 beginnt und mit dem Basenpaar 61153555 endet. In diesem Bereich, innerhalb dieser DNA-Sequenz müssen sich demnach sämtliche Promotoren, sämtliche poly-A-Signale und sämtliche ALU-Gene befinden, die für die Definition und die Verarbeitung von Primärtranskripten der metabolen Gene auch Strukturgene genannt - dieser Subdomäne auf dem plus- und dem minus-Strang212 und für das Processing ihrer Transkripte zu zellspezifischen messenger-RNAs notwendig sind.
Die meisten Transkriptionsmodelle beruhen auf der Bildung von DNA-Schleifen, die an die Matrix (scaffold) gebunden werden.
Paul und Ferl haben bereits 1998 ein Modell für das Pflanzengenom vorgeschlagen213, welches davon ausging, dass relativ große Schlaufen durch konstitutiv bindende SMAR-Elemente -sogenannte loop basement attachment regions (LBARs) - gebildet werden. Andere SMAR-Elemente, die innerhalb dieser Schlaufe lokalisiert sind, sollten unterschiedliche regulatorische Funktionen haben und nur bei der aktiven Transkription an die Kernmatrix gebunden sein. Zitat:
These results imply that the genome is not packaged by means of a random gathering of the genome into domains of indiscriminate length but rather that the genome is gathered into specific domains and that a gene consistently occupies a discrete physical section of the genome. Our proposed model is that these large organizational domains represent the fundamental structural loop domains created by attachment of chromatin to the nuclear matrix at loop basements. These loop domains may be distinct from the domains created by the matrix attachment regions that typically flank smaller, often functionally distinct sections of the genome.
Dieses Modell von Paul und Ferl stimmt weitestgehend mit der von mir beschriebenen Struktur der DNA, mit meinen eigenen Berechnungen für das humane Genom und den Schlussfolgerungen daraus überein auch Sandra Götze kommt in ihrer Dissertation214 zu dem Schluss, dass sich das Modell von Paul und Ferl auf andere Organismen übertragen lässt. Zitat:
, ...dass dieses Modell auch auf andere Organismen zutrifft. Zur Gruppe der variabel und damit nicht-konstitutiv bindenden SMAR-Elemente gehören demzufolge die Promotor- und Terminator-SMARs. Diese SMARs besitzen höchstens eine moderate Affinität zur nukleären Matrix und konkurrieren mit wesentlich stärkeren SMAR-Elementen wie dem SAR-E um ein und denselben Protein-Interaktionspartner. Eine Interaktion mit der Kernmatrix ist für Promotor- und Terminator-Fragmente nur während der Transkription notwendig, deshalb muss eine konstitutive Bindung nicht vorliegen. Kandidaten für die Gruppe der konstitutiv bindenden SMAR-Elemente und damit für die LBARs sind die in dieser Arbeit charakterisierten, stark bindenden, intergenischen DNA-Fragmente. Da sie im Gegensatz zu den sogenannten Standard-SMARs nicht in der direkten Umgebung von Genen lokalisiert sind, können die intergenischen Sequenzen gut die transkriptionsunabhängige konstante Basis der DNA-Schlaufen bilden. (Zitat Ende)
Die von Paul und Ferl (und S. Götze) LBARs genannten intergenischen Elemente heissen bei mir REMAKEs es sind SMARs der Klasse III die Chromatin-Domänen und Chromomere definieren und über Bindestellen in zellspezifisch aktivierten REMA-Boxen konstitutiv an die Kernmatrix gebunden sind. Diese Bindestellen sind während der Ontogenese in einem engen Rahmen, der durch die Grenzen des REMAKEs gesetzt wird, veränderbar in einer ausdifferenzierten Zelle dagegen konstant. Sie sind ausser mit der Transkription eng mit der DNA-Replikation und der Kompaktierung des Chromatins während der Mitose verknüpft. Darüber habe ich in Teil I und II dieser Arbeit und weiter oben bereits ausführlich referiert. Die von Paul und Ferl erwähnten SMARs, die kleinere DNA-Regionen mit spezifischen Funktionen einschließen, sind die von mir SMARS der Klasse III genannten Abschnitte vor Subdomänen, die nicht konstitutiv an der Matrix fixiert sind. Auch Promotoren und Terminatoren sowie SMARs der Klasse II, welche die Grenzen von ALU-Genen und REMA-Genen definieren, gehören zu solchen nicht konstitutiv, sondern nur temporär an die Kernmatrix bindenden DNA-Motiven.
Man sollte sich bei der Auseinandersetzung mit diesem Thema aber stets vor Augen halten, dass REMAKEs abhängig von der Phase des Zellzyklus unterschiedliche Funktionen wahrnehmen können. So agieren sie in der G0- oder Interphase als Subdomänen SMARs der Klasse III, welche die Dekondensation von Subdomänen und damit die Transkription eines Gens in dieser Subdomäne zum Beispiel der Subdomäne 3.1 aktivieren und regulieren können. Ein REMAKE (der konstitutiv an die Kernmatrix gebunden ist) verhält sich dann wie eine (temporär bindende) Subdomänen-SMAR, indem er die gleichen Proteine wie diese bindet.
Der Weg zur zellspezifischen mRNA:
Ein 7-Stufen-Modell.
Das Entpacken eines postmitotischen Chromosoms vollzieht sich in mehreren Schritten, die sämtlich mit der Bindung stammzellspezifischer Architektur-Proteine an SMARs der Klasse II und III verbunden sind. Die Stufe, auf der die zellspezifischen Architekturproteine in die Organisation des Chromatins eingreifen, stellt vermutlich das 30nm-Solenoid dar. Am letzten Schritt, der Installation einer trankriptionsfähigen Subdomäne und an der Bildung von Transkriptionsschleifen in dieser Subdomäne sind sowohl formatierungshormonspezifische HMGN-, HMGA-, SAF- und SATB-Proteine beteiligt als auch HRPKs und Proteine, die von einem primären Transkriptionshormon (SAF) und Proteine, die von einem sekundären Transkriptionshormon (ARBP, HMGB) aktiviert werden. Die entscheidenden Schritte hin zu einem zellspezifisch transkribierten Gen erfolgen demnach auf der Subdomänen-Ebene.
Dass das Entpacken eines Chromomers nicht wahl- und planlos geschieht, sondern strengen Regeln folgt, denen ein logisches und ein logistisches System zugrunde liegen, habe ich zu Beginn dieses Teils III zu zeigen versucht. Ich habe den gesamten Entpackungs- und Aktivierungsprozess deshalb in sieben Stufen eingeteilt, die ich wie folgt charakterisiere:
A215
Ausgangsbasis: das Chromomer.
B
Öffnen der Chromomer-Schleifen: die DNA als lineares 30nm-Solenoid.
C
Formierung der Subdomänen-Loops: Binden der Subdomänen-SMARs mit
Hilfe von HMGA/SATB-, HMGN- und SAF-Proteinen an die Matrix. Das Solenoid wird zur 10nm kondensierten Nukleosomenkette entspannt.
D
Öffnen der Ziel-Subdomäne durch ein zellspezifisches, von einem sekundären Transkriptionshormon aktiviertes Protein der ARBP-Familie.
Die zuvor kondensierte 10nm- Nukleosomenkette wird dekondensiert.
E
Ersatz von H1 durch den H1PAC217 einen Komplex aus einem HMGN-Protein, einem zellspezifisch aktivierten HRPK, Ubiquitin und Nukleoplasmin.
F
Bindung eines Terminators (poly-A-Stelle) an einen Promotor unter Beteiligung von zellspezifischen Architekturproteinen. Der Promotor/Terminator-Komplex wird an die SMAR/Kernmatrix gebunden.
Es entstehen eine inaktive DNA-Schleife und eine aktive Transkriptionsschleife, welche die Sequenz zwischen Promotor und Terminator umfasst.
G
Die allgemeinen Transkriptionsfaktoren, eine Helikase, eine Topo-Isomerase und die RNAPII binden an den SMAR/Promotor/Terminator-Komplex, der so zum Transkriptosom wird. Die in dieser primären Schleife kodierten UsnRNAs und spezifischen TAFs (SNIRPs und TAPs) werden transkribiert.
H
Die spezifische durch die Hormon-Triade definierte TSS (Beispiel: TRH->TSH->y-Lipotropin) wird an das Transkriptosom gebunden. Die zweite (sekundäre) Transkriptionsschleife formiert sich zwischen einer definierten TSS und dem Terminator. Transkription des Primärtranskripts, das mit Hilfe der zuvor generierten UsnRNAs, SNIRPs und TAPs zu einer zell- und entwicklungsspezifischen messenger-RNA gespleisst wird.
Wie ich an anderer Stelle schon gesagt habe, stellt die Subdomäne in Verbindung mit dem REMAKE der übergeordneten Domäne eine genetische Funktionseinheit dar, in der die essentiellen Faktoren, die für eine differentielle Genexpression erforderlich sind, konzentriert sind. Dies sind vor allem Promotoren, Terminatoren (poly-A-Signale), ALU-Gene, welche zellspezifische Transkriptionsfaktoren und UsnRNAs kodieren sowie SMARs der Klasse II und III und die von mir weiter oben charakterisierten Architekturproteine, welche die zell- und entwicklungs-abhängige Strukturierung des Chromatins in funktionelle Einheiten organisieren.
Obwohl ich diese These im Augenblick nicht beweisen kann, würde ich trotzdem wagen zu behaupten, dass ein bestimmter DNA-Strang einer Subdomäne auch immer nur ein einziges metaboles polycistronisches216 Gen enthalten kann. Dies schliesse ich nicht zuletzt aus der Tatsache, dass in einer Subdomäne eben die Faktoren kodiert sind, die für eine zell- und entwicklungsspezifische Transkription des inkludierten metabolen polycistronischen Gens erforderlich sind. Wir wollen uns deshalb diese Faktoren und die Struktur der Domäne 3 sowie die der Subdomäne 3.3 etwas genauer ansehen:
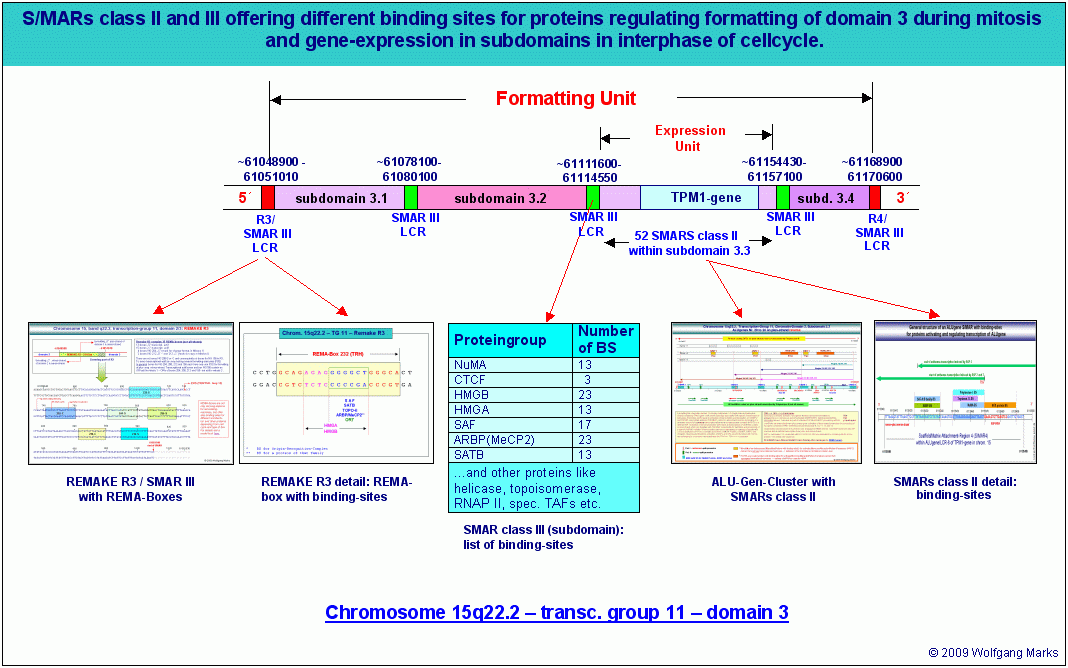
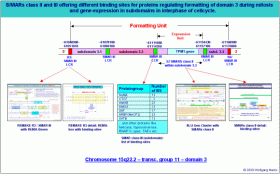 Die Abbildung gibt einen Überblick über Organisation und Struktur der Domäne 3 und insbesondere der Subdomäne 3.3 in der Transkriptionsgruppe 11 des Chromosomenbandes 15q22.2 mit Angabe der bis heute von mir identifizierten Strukturelemente und der Bindestellen für Architekturproteine in diesen Elementen. Alle hier vorgestellten Struktur-Elemente haben wir in Teil I und II dieser Arbeit bereits kennengelernt. Wir können also jetzt versuchen, am Beispiel der Domäne 3 und der Subdomäne 3.3 ein logisches Modell der differentiellen Expression polycistronischer Gene zu entwickeln. Die Abbildung gibt einen Überblick über Organisation und Struktur der Domäne 3 und insbesondere der Subdomäne 3.3 in der Transkriptionsgruppe 11 des Chromosomenbandes 15q22.2 mit Angabe der bis heute von mir identifizierten Strukturelemente und der Bindestellen für Architekturproteine in diesen Elementen. Alle hier vorgestellten Struktur-Elemente haben wir in Teil I und II dieser Arbeit bereits kennengelernt. Wir können also jetzt versuchen, am Beispiel der Domäne 3 und der Subdomäne 3.3 ein logisches Modell der differentiellen Expression polycistronischer Gene zu entwickeln.
Um das Szenario übersichtlich zu halten, gehe ich davon aus, dass sich nur in der Subdomäne 3.3 ein Gen befindet (das TPM1-Gen) dessen Expression durch das Formatierungshormon TRH und das primäre Transkriptionshormon TSH aktiviert werden kann. Wäre das anders (was ich nicht geprüft habe), müssten die Prozesse, die ich für die Subdomäne 3.3 schildere, bis zur Einleitung der TSS-gesteuerten Transkription auch für die Subdomäne gelten, die ein solches Gen enthält.
Für den Weg zur Transkriptionsschleife, den ich hier am Beispiel des TPM1-Gens beschreibe, lassen sich in dem einen oder anderen Stadium auch Alternativen denken. Allen Alternativen aber sollte gemeinsam sein, dass jeder Schritt hin zur zellspezifischen messenger-RNA mit einer kontinuierlichen Einengung des Aktionspotentials der Zelle verbunden ist. Dieser Weg Vom Allgemeinen hin zum Besonderen ist an die Ausschüttung von Hormonen der Hormonkaskade, an die Bindung von Architekturproteinen in SMARs und schliesslich an die Synthese zell- und entwicklungsspezifischer Transkriptionsfaktoren gekoppelt, welche durch die Hormone der Hormonkaskade koordiniert aktiviert werden.
Der Weg zur zellspezifischen mRNA: Das Chromomer.
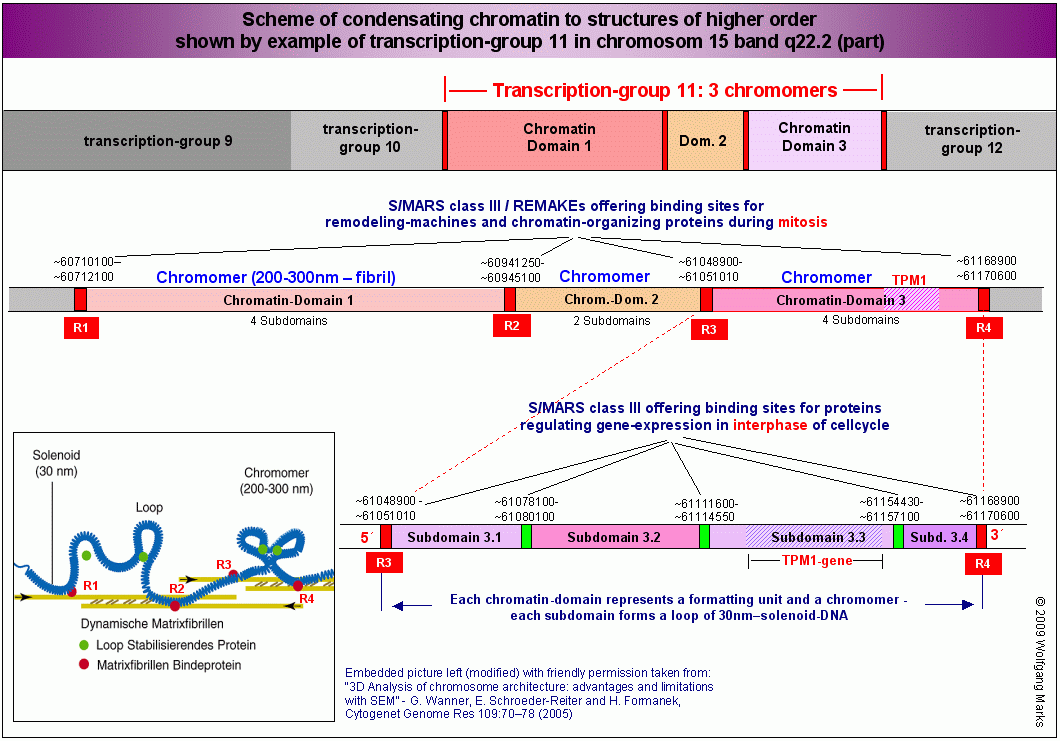
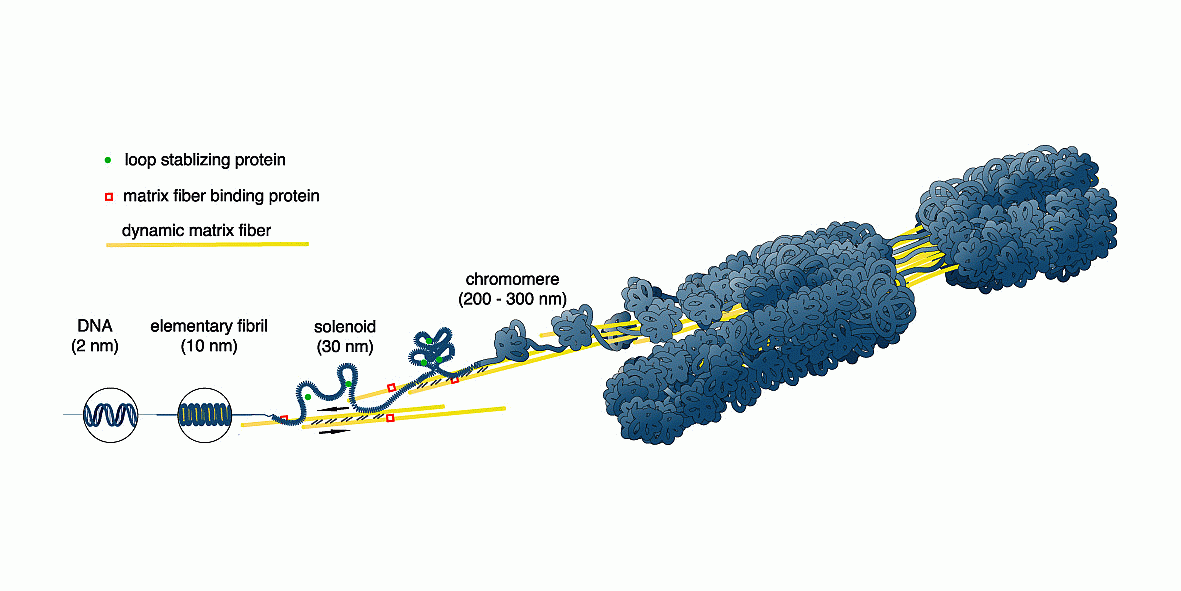
Nach der Zellteilung liegt die DNA zunächst als Chromomer vor. Dieses besteht aus einer 30nm-Solenoid-Fiber, deren Windungen sich in Abhängigkeit von der Nukleosomengrösse aus 6 bis 8 Nukleosomen zusammensetzen. Die Solenoid-Struktur wird zum einen durch Kontakte zwischen den H1-Histonen, zum anderen wohl auch durch Torsionsspannungen aufrecht erhalten, die bei der Bildung der Chromomer-Schleifen durch Kontraktion von Kernmatrix-Fibern entstanden sind (siehe hierzu auch das Chromosomenmodell von Wanner und Formanek).
 Die SMARs an beiden Enden der Domäne sind REMAKEs konstitutiv an die Matrix gebundene DNA-Regionen, die Bindestellen für eine Reihe von Architekturproteinen bereithalten. Welche Proteine hier gebunden werden, ist sowohl von der Zellzyklus-Phase, als auch vom Entwicklungsschritt abhängig, in dem sich die Zelle befindet. Proteine der SATB-Familie zum Beispiel binden hier erst ab dem 110. Tag der Ontogenese (Beginn der differentiellen Genexpression) das gleiche gilt für Proteine der SAF-Familie. Auch CTCF-Proteine binden in REMAKEs von einem bestimmten Zeitpunkt (80. Tag) an aber nur dann, wenn sich in der Domäne ein Gen befindet, das dem Imprinting unterliegt. Vor dem 110. Tag der Ontogenese finden wir aus der Gruppe der Architekturproteine ausschliesslich HMGA- und HMGN-Proteine in den REMAKEs von Chromomeren. Die SMARs an beiden Enden der Domäne sind REMAKEs konstitutiv an die Matrix gebundene DNA-Regionen, die Bindestellen für eine Reihe von Architekturproteinen bereithalten. Welche Proteine hier gebunden werden, ist sowohl von der Zellzyklus-Phase, als auch vom Entwicklungsschritt abhängig, in dem sich die Zelle befindet. Proteine der SATB-Familie zum Beispiel binden hier erst ab dem 110. Tag der Ontogenese (Beginn der differentiellen Genexpression) das gleiche gilt für Proteine der SAF-Familie. Auch CTCF-Proteine binden in REMAKEs von einem bestimmten Zeitpunkt (80. Tag) an aber nur dann, wenn sich in der Domäne ein Gen befindet, das dem Imprinting unterliegt. Vor dem 110. Tag der Ontogenese finden wir aus der Gruppe der Architekturproteine ausschliesslich HMGA- und HMGN-Proteine in den REMAKEs von Chromomeren.
Die Schleifen des Chromomers werden bis zum Beginn der differentiellen Genexpression ebenfalls ausschliesslich durch HMGA-Proteine, danach wie gezeigt durch Proteine der SAF-, der HMGA- und der SATB-Familie zusammmengehalten, die ebenso wie die Proteine, welche das Chromomer an der Kernmatrix verankern, mit der Nukleosomengrösse der Domäne und den Formatierungshormonen, welche diese Formatierung initiiert haben, sowie primären Transkriptionshormonen korreliert sind, die von den übergeordneten Formatierungshormonen aktiviert wurden. Bei der Bildung der Chromomer-Schleifen könnte neben der Kontraktion der Kernmatrixfibern das sogenannte mass-binding, das für SAF- Proteine beschrieben wurde (Kipp et al. 2000) eine Rolle spielen.
Die Abbildung A zeigt die zuvor charakterisierte Domäne 3 mit ihren vier Subdomänen 3.1 bis 3.4 zum Chromomer kondensiert. Sie ist mit der Nukleosomengrösse 232 formatiert. Das Chromomer selbst besteht aus zwei Schleifen (loops) die über die REMAKEs der Subdomänen 3.1 bzw. 3.4 an der Kernmatrix befestigt sind. An die REMAKEs R3 und R4 haben Architekturproteine gebunden, die mit dem Formatierungshormon TRH korreliert sind (SATB-232, HMGA-232, HMGN-232) sowie ein Molekül SAF232-TSH, dessen Synthese vom TRH-induzierten primären Transkriptionshormon TSH induziert wurde.
CTCF bindet wie am Ende von Teil II ausführlich beschrieben - nur dann in einer SMAR der Klasse III, wenn sich in einer der Subdomänen ein imprintetes metaboles Gen befindet. Das TPM1-Gen, dessen Expression von nun an im Mittelpunkt des Interesses stehen wird, unterliegt allerdings nicht dem Imprinting.
Das Untersuchungsobjekt: TPM1.
Zitat (NCBI RefSeq):This gene is a member of the tropomyosin family of highly conserved, widely distributed actin-binding proteins involved in the contractile system of striated and smooth muscles and the cytoskeleton of non-muscle cells. Tropomyosin is composed of two alpha-helical chains arranged as a coiled-coil. It is polymerized end to end along the two grooves of actin filaments and provides stability to the filaments. The encoded protein is one type of alpha helical chain that forms the predominant tropomyosin of striated muscle, where it also functions in association with the troponin complex to regulate the calcium-dependent interaction of actin and myosin during muscle contraction. In smooth muscle and non-muscle cells, alternatively spliced transcript variants encoding a range of isoforms have been described. Mutations in this gene are associated with type 3 familial hypertrophic cardiomyopathy.
Das TPM1-Gen ist ein aus 15 Exons bestehendes polycistronisches Gen: seine Expression beginnt etwa am 80. Tag mit dem Auftreten des Hormons Thymosin ß-10 (NG 216). Meine Aussage, dass ein polycistronisches Gen auch schon vor dem 110. Tag der Ontogenese also vor dem Beginn des differentiellen oder alternativen Spleissens exprimiert wird, ist zunächst überraschend, scheint bei einigem Nachdenken aber logisch. Denn das alternative Spleissen hat sich mit ziemlicher Sicherheit parallel zum splitting der ursprünglich einmal homogenen, aus einer ununterbrochenen DNA-Sequenz bestehenden Urgene entwickelt. Bevor das alternative Spleissen im Verlauf der Phylogenese entwickelt wurde (für das neben den UsnRNAs eine Reihe spezifischer Proteine benötigt werden), hat es deshalb mit grosser Wahrscheinlichkeit bereits ein rudimentäres lineares (sequentielles) Spleissen von Exons unter Beteiligung von UsnRNAs gegeben. Auf die Ontogenese bezogen, dürfte dieser Zeitpunkt mit dem Beginn der Fetogenese zusammenfallen lineares oder sequentielles Spleissen von polycistronischen Genen tritt demnach etwa ab dem 60. Tag der Ontogenese auf.
Alternatives Spleissen aber findet vor dem 110. Tag der Ontogenese mit ziemlicher Sicherheit nicht statt, da weder die dazu benötigten Proteine, noch die ebenfalls dazu benötigten sekundären (processsing)-Hormone zur Verfügung stehen.
Die uns hier interessierende TPM1-messenger-RNA, die aus einer TRH-induzierten, TSH-abhängigen hnRNA unter dem Einfluss von y-Lipotropin gespleisst wird, wird etwa vom 112. Tag der Ontogenese an generiert. Der Weg, den die Zelle beschreitet, um dieses spezifische Transkript zu einem definierten Zeitpunkt in einer definierten Zelle zu exprimieren, soll jetzt Schritt für Schritt untersucht werden.
Der Weg zur zellspezifischen mRNA: Das Öffnen der Chromomer-Schleifen.
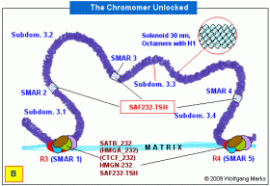 Das Öffnen der Chromomer-Schleifen ist vermutlich eine Folge der chemischen Modifizierung von SATB-232 und HMGA-232. Diese Modifizierungen bestehen vermutlich aus Methylierungen und Demethylierungen auf der einen sowie Phosphorylierungen und Dephosphorylierungen auf der anderen Seite. Ich nehme an, dass SATB-232 an bestimmten Positionen phosphoryliert und methyliert, HMGA-232 vermutlich ebenso spezifisch demethyliert und dephosphoryliert wird. Beide Proteine dissoziieren als Folge der Modifikationen von der Solenoid-DNA, die damit nur noch über die REMAKEs R3 und R4 an der Kernmatrix befestigt ist. SAF232-TSH bleibt an die Subdomänen-SMARs gebunden. Die Konformation und die Zusammensetzung des Proteinkomplexes an den REMAKEs R3 und R4 ändert sich in diesem Stadium nicht. Das Öffnen der Chromomer-Schleifen ist vermutlich eine Folge der chemischen Modifizierung von SATB-232 und HMGA-232. Diese Modifizierungen bestehen vermutlich aus Methylierungen und Demethylierungen auf der einen sowie Phosphorylierungen und Dephosphorylierungen auf der anderen Seite. Ich nehme an, dass SATB-232 an bestimmten Positionen phosphoryliert und methyliert, HMGA-232 vermutlich ebenso spezifisch demethyliert und dephosphoryliert wird. Beide Proteine dissoziieren als Folge der Modifikationen von der Solenoid-DNA, die damit nur noch über die REMAKEs R3 und R4 an der Kernmatrix befestigt ist. SAF232-TSH bleibt an die Subdomänen-SMARs gebunden. Die Konformation und die Zusammensetzung des Proteinkomplexes an den REMAKEs R3 und R4 ändert sich in diesem Stadium nicht.
Der Weg zur zellspezifischen mRNA: Die Bildung der Subdomänen-Loops.
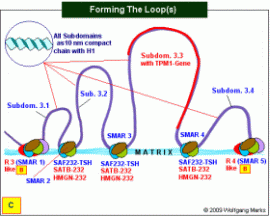 An die SAF232-TSH-Moleküle, die an den SMARs 2 bis 4 verblieben sind, bindet zunächst je ein Molekül HMGN-232, das einen TSH-HRPK gebunden hat. Danach bindet der Komplex an die Kernmatrix, wo er durch SATB-232 vervollständigt wird. Durch die Bindung der genannten Proteine und die Fixierung der DNA an der Matrix entspannt sich das 30nm-Solenoid zur 10nm kondensierten Nukleosomenkette mit gebundenem H1. Damit ist die Domäne 3 in vier unterschiedlich grossen Schleifen, die jeweils eine Subdomäne repräsentieren, mit ihren SMARS der Klasse III an der Kernmatrix fixiert. An die SAF232-TSH-Moleküle, die an den SMARs 2 bis 4 verblieben sind, bindet zunächst je ein Molekül HMGN-232, das einen TSH-HRPK gebunden hat. Danach bindet der Komplex an die Kernmatrix, wo er durch SATB-232 vervollständigt wird. Durch die Bindung der genannten Proteine und die Fixierung der DNA an der Matrix entspannt sich das 30nm-Solenoid zur 10nm kondensierten Nukleosomenkette mit gebundenem H1. Damit ist die Domäne 3 in vier unterschiedlich grossen Schleifen, die jeweils eine Subdomäne repräsentieren, mit ihren SMARS der Klasse III an der Kernmatrix fixiert.
Der Weg zur zellspezifischen mRNA: Die Öffnung der Nukleosomenkette.
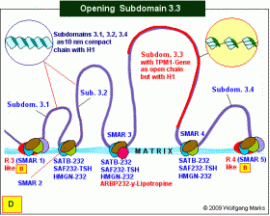 Das durch TRH/TSH aktivierte sekundäre Transkriptionshormon y-Lipotropin induziert die Synthese des Architekturproteins ARBP232-y-Lipotropin, das an seine spezifischen Bindestelle in der SMAR3 bindet. Dieser Bindung geht wahrscheinlich eine spezifische Modifikation der DNA voraus, die im Falle von ARBP232-y-Lipotropin vermutlich aus einer Methylierung von C- und G-Nukleotiden, verbunden mit einer Phosphorylierung der Adenin-Bausteine besteht. (Wenn das an die Subdomänen-SMAR bindende Protein MeCP2 (ARBP252-??) wäre, würde die analoge Modifikation der DNA in einer Demethylierung von CpG-Inseln bestehen.) Als Folge der Bindung von ARBP232-y-Lipotropin und der Modifikation der DNA lösen sich definierte Bindungen zwischen den H1-Histonen und der DNA die kompakte Nukleosomenkette der Ziel-Subdomäne 3.3 öffnet sich zur offenen Nukleosomenkette mit dem spacing 232. H1 bleibt aber vorerst noch an die Oktamere fixiert. Das durch TRH/TSH aktivierte sekundäre Transkriptionshormon y-Lipotropin induziert die Synthese des Architekturproteins ARBP232-y-Lipotropin, das an seine spezifischen Bindestelle in der SMAR3 bindet. Dieser Bindung geht wahrscheinlich eine spezifische Modifikation der DNA voraus, die im Falle von ARBP232-y-Lipotropin vermutlich aus einer Methylierung von C- und G-Nukleotiden, verbunden mit einer Phosphorylierung der Adenin-Bausteine besteht. (Wenn das an die Subdomänen-SMAR bindende Protein MeCP2 (ARBP252-??) wäre, würde die analoge Modifikation der DNA in einer Demethylierung von CpG-Inseln bestehen.) Als Folge der Bindung von ARBP232-y-Lipotropin und der Modifikation der DNA lösen sich definierte Bindungen zwischen den H1-Histonen und der DNA die kompakte Nukleosomenkette der Ziel-Subdomäne 3.3 öffnet sich zur offenen Nukleosomenkette mit dem spacing 232. H1 bleibt aber vorerst noch an die Oktamere fixiert.
Der Weg zur zellspezifischen mRNA: der Ersatz von H1 durch den H1PAC. Die Subdomäne wird transkriptionsfähig.
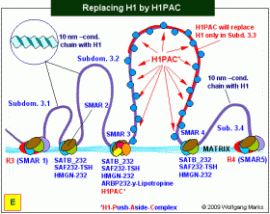 Damit ein Gen transkribiert werden kann, müssen das habe ich bereits mehrmals ausführlich diskutiert die aktivierenden oder regulierenden Proteine an spezifische DNA-Motive binden können. Dazu muss die DNA als dekondensierte 10nm-Fiber vorliegen, also als geöffnete Nukleosomenkette mit einem der Formatierung entsprechenden spacing: im konkreten Fall ist das die NG 232. Damit ein Gen transkribiert werden kann, müssen das habe ich bereits mehrmals ausführlich diskutiert die aktivierenden oder regulierenden Proteine an spezifische DNA-Motive binden können. Dazu muss die DNA als dekondensierte 10nm-Fiber vorliegen, also als geöffnete Nukleosomenkette mit einem der Formatierung entsprechenden spacing: im konkreten Fall ist das die NG 232.
Aber auch in dieser Form wäre das TPM1-Gen nicht transkribierbar, weil eine Nukleosomenkette, deren Cores H1 gebunden haben, nach allgemeiner Auffassung nicht abgelesen werden kann. Es läge also nahe anzunehmen, dass die Subdomäne 3.3 im Augenblick der Transkriptionsaktivierung entweder völlig frei von H1 ist (was durch eine entsprechende Modifizierung der H1-Histone während der vorangehenden Mitose erreicht werden könnte) oder dass die H1-Histone einzig von dem Teil der DNA verdrängt werden, der transkribiert werden soll und in den anderen Abschnitten persistieren. An dieser Stelle möchte ich daran erinnern, dass sowohl die DNA, als auch die Histone des Oktamers in der Chromatin-Domäne 3.3 in jeder Phase der Ontogenese zellspezifisch derart modifiziert sind, dass die nukleosomalen Einheiten aus einem Oktamer und H1 an definierte Positionen in der DNA binden. Eine Ablösung der H1-Histone von der DNA und den Oktameren hätte also mit Sicherheit die Instabilität der DNA-Oktamer-Bindungen zur Folge eine Instabilität, die entweder zum wobbling218 der Oktamere auf der DNA oder zur Deassemblierung der Oktamere führen würde. Beide Phänomene würden eine zielgerichtete Bindung von Transkriptionsfaktoren unmöglich machen. H1 kann also nicht entfernt werden, ohne durch ein adäquates Surrogat ersetzt zu werden.
Nach allem, was von Voruntersuchern über die Eigenschaften und das Verhalten von HMGN-Proteinen berichtet wurde, kommt von den zuvor charakterisierten Architekturproteinen für die Aufgabe, H1 adäquat zu ersetzen, nur HMGN in Betracht. Sowohl in vitro als auch in vivo konnte nachgewiesen werden, dass sowohl HMGN1 als auch HMGN2219 eine hohe Affinität zu Nukleosomen aufweisen: sie binden in vitro gezielt und proteinspezifisch an definierte zusammenhängende Chromatinbereiche. Vor allem die Arbeiten von R. Hock und M. Bustin haben gezeigt, dass die experimentelle Verdrängung von HMGN2 zu einer adäquaten Verringerung der Transkriptionsrate führt bei vollständiger Hemmung der Transkription ist das Protein nahezu vollständig in den Interchromatin-Granula konzentriert.
Dass die Bindung von HMGN-Proteinen an das Chromatin essentiell für die Expression zell- und entwicklungsspezifischer Gene ist, konnte Ulrich Körner aus der Arbeitsgruppe um R. Hock in seiner zuvor bereits erwähnten Arbeit über die funktionelle Rolle der HMGN-Proteine während der Embryogenese des südafrikanischen Krallenfrosches (Xenopus laevis) nachweisen. Er kam zu dem Ergebnis, dass sowohl die Expression als auch die zelluläre Verteilung der HMGN-Proteine entwicklungs-spezifisch reguliert wird. Das Auftreten von HMGN-Proteinen im Zellkern ist nach dem Ergebnis seiner Untersuchungen eng mit der transkriptionellen Aktivierung des embryonalen Genoms verknüpft: obwohl während der Oogenese in allen Oozytenstadien zu finden, verschwinden HMGN-Proteine während der Maturation der Xenopus-Oozyten zu Eiern und sind erst mit dem Beginn der Embryogenese und dort ab dem Blastula-Stadium - wieder nachweisbar. Dabei weisen ihre Expressionsmuster laut Körner zumindest auf mRNA-Ebene auf Gewebespezifität hin.
Körners Analysen zeigen auch, dass eine bestimmte kritische zelluläre HMGN- Proteinmenge für eine korrekte Embryonalentwicklung von Xenopus laevis notwendig ist. Durch animal cap assays und RT-PCR-Expressionsanalysen Mesoderm-spezifischer Gene konnte er nachweisen, dass HMGN-Proteine die Regulation mesoderm-spezifischer Gene beeinflussen. Durch eine Analyse des Expressionsbeginns entwicklungsrelevanter Gene während der Midblastula-Transition konnte er ausserdem belegen, dass veränderte HMGN- Proteinmengen den Expressionsbeginn spezifischer Xenopus-Gene wie Xbra und chordin beeinflussen. Damit konnte er erstmals einen direkten Zusammenhang zwischen der Expression von HMGN-Proteinen und der Expression spezifischer Gene herstellen.
Lim et al. (2002) postulieren, dass ein Großteil der HMGN-Proteine in makromolekularen Komplexen organisiert ist: in HeLa-Zellen fanden sie sowohl HMGN1 als auch HMGN2 in
größeren metastabilen Komplexen, deren andere Komponenten aber nicht identifiziert werden konnten. Wurde die Transkription in den Zellen inhibiert, erhöhte sich die Anzahl der freien, ungebundenen HMGN-Moleküle und vice versa. Die Inkorporation in makromolekulare Komplexe ist also offensichtlich mit der transkriptionellen Aktivität der Zelle korreliert. Lim et al. konnten darüberhinaus nachweisen, dass die Bindungsaffinität zwischen komplexgebundenen HMGN-Proteinen und Nukleosomencores größer ist als die zwischen ungebundenen HMGN-Proteinen und Corepartikeln.
Dass HMGN-Proteine als makromolekulare Komplexe mit dem Chromatin interagieren, weiss man also schon seit geraumer Zeit wie sie das tun, ist allerdings unklar.
HMGN-Proteine bilden makromolekulare Komplexe.
Sie weisen verschiedene Bindestellen für aktivierte HRPKs auf.
Ausserdem je eine Bindestelle für Nukleoplasmin und Ubiqutin.
Wenn man die zuvor zitierten Untersuchungsergebniss kritisch würdigt, liegt es nahe anzunehmen, dass H1 in der Subdomäne 3.3 noch vor der Bildung des Transkriptosoms durch einen makromolekularen Komplex verdrängt wird, der HMGN und gegebenenfalls weitere Proteine enthält. Diesen putativen Komplex habe ich H1PAC genannt. Er setzt sich (im konkreten Fall) wahrscheinlich zusammen aus einem Molekül HMGN-232, an das ein aktiviertes TSH-HRPK gebunden ist, einem Molekül Nukleoplasmin und einem Molekül Ubiquitin. Worauf sich meine Überlegungen stützen, will ich nun versuchen darzulegen:
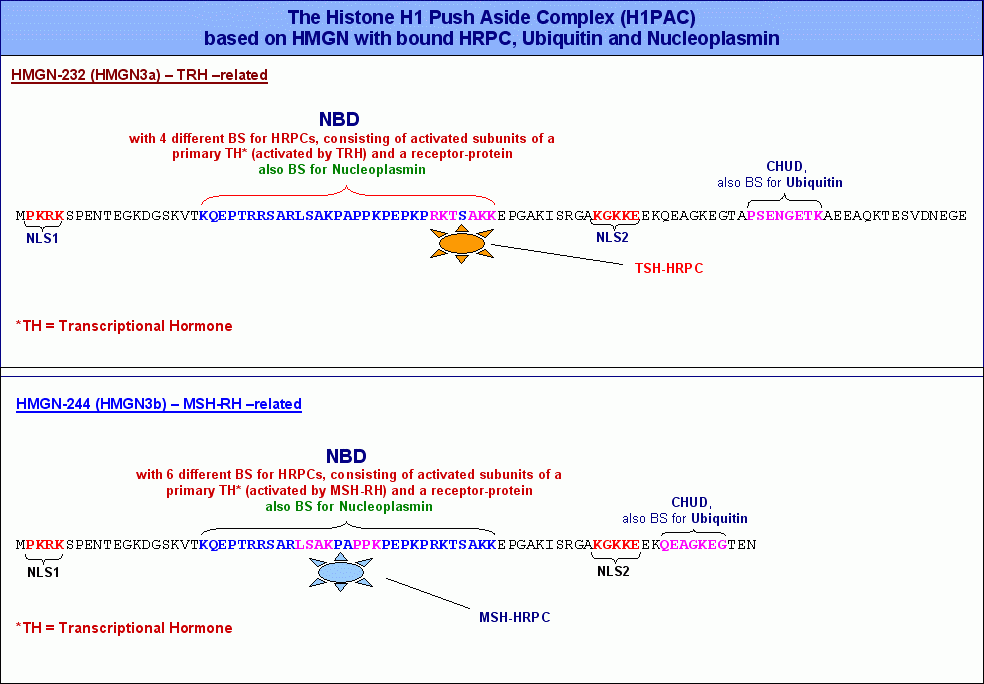
Es ist seit längerem bekannt, dass HMGN-Proteine über (zumindest) drei funktionelle Domänen verfügen: ein zweigeteiltes Kernlokalisations-Signal (nuclear localisation signal NLS), eine nukleosomale Bindungsdomäne (nucleosome-binding-domain NBD) und eine sogenannte Chromatin-Aktivierungsdomäne (chromatin unfolding domain CHUD). Diese drei Domänen sind sowohl innerhalb der HMG-Proteingruppe selbst als auch in verschiedenen Spezies mehr oder weniger hoch konserviert.
Abbildung: Bustin, 2001
Das Kernlokalisations-Signal ermöglicht es den HMGN-Proteinen, über einen aktiven Importin-abhängigen Transport rasch in den Zellkern zu gelangen (Hock et al., 1998).
Die Chromatin-Aktivierungsdomäne besteht aus einer negativ geladenen Region, die den HMGN-Proteinen die Fähigkeit verleihen soll, das Chromatin aufzulockern, indem die C-terminale Region, die die Aktivierungsdomäne enthält, mit dem aminoterminalen Bereich von H3 interagiert (Trieschmann et al., 1998). Alfonso et al. (1994) konnten zeigen, dass der Kontaktbereich zwischen HMGN1- bzw. HMGN2-Proteinen und den Nukleosomen mit dem von H1 teilweise identisch ist. Schon Bustin vermutete deshalb 1999, dass dadurch die Bindung von H1 an das Core-Partikel gestört und das Chromatin in der Folge aufgelockert wird. Catez et al. konnten 2002 schließlich demonstrieren, dass HMGN-Proteine und H1 in vivo dynamisch um die gleichen Bindungsstellen konkurrieren.
Die nukleosomale Bindungsdomäne (NBD) schliesslich besteht bei den bis jetzt bekannten HMGN-Proteinen aus 30 positiv geladenen Aminosäuren und ist zwar stark konserviert, aber nicht bei allen HMGN-Proteinen gleich. Sie soll die Struktur des nukleosomalen Core-Partikels erkennen (bestehend aus dem Histon-Oktamer und 146 bp DNA) allerdings sind für eine korrekte Bindung an die Nukleosomen wie Versuche mit Punktmutationen gezeigt haben wohl auch benachbarte Aminosäuren oder zusätzliche Faktoren erforderlich.
H1PAC ersetzt H1.
Diese zusätzlichen Faktoren sind mit grosser Wahrscheinlichkeit Ubiquitin und Nukleoplasmin, zwei Proteine, die bei der Transkriptionsaktivierung besondere Aufgaben haben, und im konkreten Fall ein zellspezifischer Komplex aus einem Rezeptorprotein und dem Hormon TSH (TSH-HRPK).
Im HMGN-232220 Protein, dessen Synthese in der aktuellen Zelle durch das Formatierungshormon TRH aktiviert wird, habe ich mittels IMPACD® ausser den Bindestellen für Ubiquitin und Nukleoplasmin insgesamt vier verschiedene Motive identifiziert, an welche HRPKs binden können, die durch die Aktivierung eines zellulären Rezeptores durch ein primäres von TRH abhängiges Transkriptionshormon entstanden sind (primäre HRPKs).
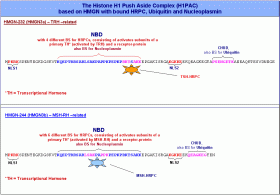 Die nebenstehende Abbildung zeigt sowohl die Aminosäuresequenz von HMGN-232, als auch die von HMGN-244, während HMGN-232 vier Bindestellen für primäre HRPKs aufweist, sollte HMGN-244 in der Lage sein, sechs verschiedene HRPKs zu binden, die jeweils aus einem von MSH-RH abhängigen primären Transkriptionshormon (zum Beispiel MSH) und einem aktivierten Rezeptorprotein bestehen. Dementsprechend sollte es auch vier von TRH und sechs von MSH-RH abhängige primäre Transkriptionshormone geben. Ich bin mir darüberhinaus sicher, dass alle HMGN-Proteine nicht nur aktivierte HRPKs binden können, sondern über die Chromatin Unfolding Domain (CHUD) auch Ubiquitin sowie über die nukleosomeale Bindungsdomäne (NBD) schliesslich das Protein Nukleoplasmin. Beide Proteine haben wie wir noch sehen werden wichtige Aufgaben beim eigentlichen Transkriptionsgeschehen. Deswegen werde ich die Rolle, die diese beiden Proteine bei der Transkription spielen, erst bei der Besprechung des Transkriptosoms und des Transkriptionsablaufs diskutieren. Die nebenstehende Abbildung zeigt sowohl die Aminosäuresequenz von HMGN-232, als auch die von HMGN-244, während HMGN-232 vier Bindestellen für primäre HRPKs aufweist, sollte HMGN-244 in der Lage sein, sechs verschiedene HRPKs zu binden, die jeweils aus einem von MSH-RH abhängigen primären Transkriptionshormon (zum Beispiel MSH) und einem aktivierten Rezeptorprotein bestehen. Dementsprechend sollte es auch vier von TRH und sechs von MSH-RH abhängige primäre Transkriptionshormone geben. Ich bin mir darüberhinaus sicher, dass alle HMGN-Proteine nicht nur aktivierte HRPKs binden können, sondern über die Chromatin Unfolding Domain (CHUD) auch Ubiquitin sowie über die nukleosomeale Bindungsdomäne (NBD) schliesslich das Protein Nukleoplasmin. Beide Proteine haben wie wir noch sehen werden wichtige Aufgaben beim eigentlichen Transkriptionsgeschehen. Deswegen werde ich die Rolle, die diese beiden Proteine bei der Transkription spielen, erst bei der Besprechung des Transkriptosoms und des Transkriptionsablaufs diskutieren.
Ob sich der H1PAC vor der Bindung an die Nukleosomen-Cores im Karyoplasma formiert oder sukzessive an den Cores und der DNA aufbaut, kann dahingestellt bleiben. Der Effekt ist der gleiche: H1 wird aus seiner Position verdrängt Damit sind die Subdomäne 3.3 und das TPM1-Gen von nun an transkribierbar.
|


{kind=link}
{kind=link}