|
|
 |
|
|
|
Wolfgang Marks: Die Formatierte DNA
|
|
|
Die epigenetische Formatierungskaskade und der
differentielle Zellzyklus.
Wie entstehen epigenetisch vererbbare Chromatinstrukturen?
Dass es einen oder mehrere Prozesse geben muss, durch die ein Netzwerk von epigenetischen Informationen errichtet und von Zelle zu Zelle und von Organismus zu Organismus weitergegeben wird, kann wohl nicht bestritten werden. Worauf dieses Netzwerk beruht und welche Mechanismen oder Faktoren es im einzelnen etablieren, war bis zur Veröffentlichung dieser Arbeit nicht gewußt.
Ich hoffe deutlich gemacht zu haben, dass es ein Netzwerk von Proteinen gibt, die durch antisense-Transkripte von REMA-Genen kodiert und durch UsnRNAs, die auf dem sense-Strang derselben Gene kodiert sind, gespleisst werden. Ich habe in einem Modell gezeigt, wie diese Proteine remodeling-Faktoren - sich zu remodeling-Maschinen zusammenschließen und zellspezifisch nukleosomale Strukturen installieren.
Ich habe gezeigt, dass die Aktivierung dieser Gene durch Promotoren erfolgt, deren Zugänglichkeit und damit Aktivierung wiederum von der Formatierung bestimmter DNA-Abschnitte (Chromatin-Domänen) mit einer definierten Nukleosomengrösse abhängig ist.
Epigenetisches chromatin-remodeling erfolgt also nach dem Ergebnis meiner Untersuchungen nicht posttranslational, und auch im eigentlichen Sinne nicht prätranskriptionell, sondern wie ich es nenne - institutionell auf einer Ebene, die der Transkription und Translation vorgelagert oder besser überlagert ist. Ich würde es einen Informationslayer nennen einen Layer, der aus REMA-Genen (ORFs) besteht, deren Promotoren wie weiter oben gezeigt - strangweise über eine gemeinsame Nukleosomengrösse netzwerkartig miteinander verknüpft sind und deren koordinierte Aktivierung zur Generierung von Proteinen führt, die sich zu ATP-abhängigen remodeling-Maschinen132 zusammenschliessen.
Die derart aktivierten Remodeling-Komplexe sind die tatsächlichen und alleinigen Motoren und Effektoren von epigenetischen (vererbbaren) Chromatin-Strukturen. So verstanden geht epigenetisches remodeling also der Transkription und Translation voraus: es setzt die Bedingungen, unter denen Genexpression, Proliferation und Differenzierung während der Ontogenese stattfinden.
Epigenetisches remodeling basiert auf der Formatierung
von Chromatin-Domänen mit prädestinierten (programmierten)
zellspezifischen Nukleosomengrössen.
Grundsätzlich hat die Zelle, wenn sie aus einem Zustand 1 in einen Zustand 2 wechseln will, drei verschiedene Möglichkeiten, mit definierten Abschnitten der DNA umzugehen:
1. Wenn sie den Informationsgehalt eines Abschnitts übernehmen will,
muss die Formatierung dieses Abschnitts kopiert werden.
2. Wenn sie den Informationsgehalt eines Abschnitts verändern will,
muss dieser Abschnitt neu formatiert werden.
3. Wenn sie den Informationsgehalt eines Abschnitts löschen will,
muss dieser Abschnitt so formatiert werden, dass auf die die Information
nicht mehr zugegriffen werden kann.
Oder in Nukleosomengrössen ausgedrückt:
1.Chromatin-Domänen, deren Formatierung übernommen werden soll, werden mit der Nukleosomengrösse 212 formatiert (Format der Domäne kopieren)
2.Chromatin-Domänen, deren Formatierung verändert werden soll, werden mit der Nukleosomengrösse 204 formatiert (Domäne neu formatieren).
3.Chromatin-Domänen, deren Formatierung gelöscht soll, werden mit der Nukleosomengrösse 208 formatiert (Domäne stillegen/inaktivieren).133
Bei einer proliferativen Mitose wird die Zelle nur Option 1 nutzen: die komplette Formatierung des Genoms, die Nukleosomenstruktur von mir Genomformat134 genannt wird kopiert und damit unverändert an die Nachkommen weitergegeben. Das Expressionsmuster der Tochterzellen verändert sich dabei nicht.
Dieser Prozess läuft wahrscheinlich in einer einzigen Mitose ab: in der S-Phase werden die Histone nach dem Muster der templates zunächst kopiert - in der G2-Phase von den zuvor ebenfalls in der G2-Phase synthetisierten remodeling-Maschinen wieder zellspezifisch modifiziert.
Die Differenzierung der gleichen Zelle jedoch erfordert einen Umbau des Chromatins: einige Bereiche des Chromatins müssen unverändert repliziert, andere Bereiche stillgelegt, wieder andere in der Struktur verändert werden.
Differenzierung erfordert zwei Zellzyklen.
Differenzierung ist ein komplexer zellulärer Prozess, der an verschiedene interne und externe Bedingungen geknüpft ist. Welche Informationen, welche Regelwerke diesen Prozess steuern, war bisher nur ansatzweise bekannt. Auch die Frage, ob die Differenzierung einer Zelle in einem Schritt (einer Mitose) ablaufen kann oder ob dazu deren zwei erforderlich sind (also zwei Mitosen), ist noch in der Diskussion.
Wenn man die Literatur der letzten Jahre zum Thema Histon-Varianten und Chromatin-Remodeling sichtet (was mir auch nicht annäherungsweise komplett gelungen ist zu umfangreich sind die Texte zu diesen Themen) fällt aber auf, dass offensichtlich stillschweigend davon ausgegangen wird, dass die mit chromatin-remodeling verknüpften Phänomene auf einen singulären Zellzyklus beschränkt sind. Dies mag damit zusammenhängen, dass die Zellkulturen, mit denen gearbeitet wird, in der Regel immortalisierte Zell-Linien (sehr oft HeLa-Zellen) sind, die zwar noch proliferieren, aber sich nicht weiter differenzieren können. Die Abläufe und Prozesse, über die hier gesprochen werden soll, unterscheiden sich in proliferierenden Zellen aber grundsätzlich von denen in Zellen, die das Stadium der Differenzierung durchlaufen, welches in diesem Zusammenhang natürlich von besonderem Interesse ist.
Das Modell einer differentiellen Mitose in zwei Zellzyklen, aber nur einer Zellteilung, welches ich anschliessend vorstelle, beruht zum einen auf einer Fülle von Beobachtungen von Voruntersuchern (die nicht immer konfliktfrei zu interpretieren sind), zum anderen auf logischen Überlegungen und Schlußfolgerungen, die sich zwingend aus diesen Beobachtungen, meinen eigenen Berechnungen und dem Konzept Die Formatierte DNA ergeben. Auf der Basis dieses Zwei-Zellzyklen-Modells wie ich es nenne lassen sich viele, wenn nicht alle Ergebnisse meiner Voruntersucher, die sich zum Teil widersprechen, nicht nur zwanglos und einleuchtend, sondern auch stringent und logisch erklären.
Differenzierung ist epigenetisch programmiert.
Wenn wir als gegeben annehmen, dass die Formatierung von Chromatin-Domänen mit definierten, distinkten Nukleosomengrössen die genomischen Bedingungen setzt, innerhalb derer differentielle Genexpression allein stattfinden kann, sind die Schlussfolgerungen, die sich daraus für den Ablauf einer differentiellen Mitose ergeben, evident:
Epigenetischem Remodeling in einem zweiten Zellzyklus muss ein normatives remodeling, also ein Markieren der geplanten Veränderungen in einem ersten Zellzyklus vorausgehen. Während das Kopieren von epigenetischen Informationen zum Zwecke der Proliferation in einem einzigen Zellzyklus ablaufen kann, lassen sich die beiden zuvor genannten Prozesse also das Markieren der vorgesehenen Veränderungen und die Ausführung derselben - bei einer sich differenzierenden Zelle aus naheliegenden Gründen nicht in einem einzigen Zellzyklus unterbringen:
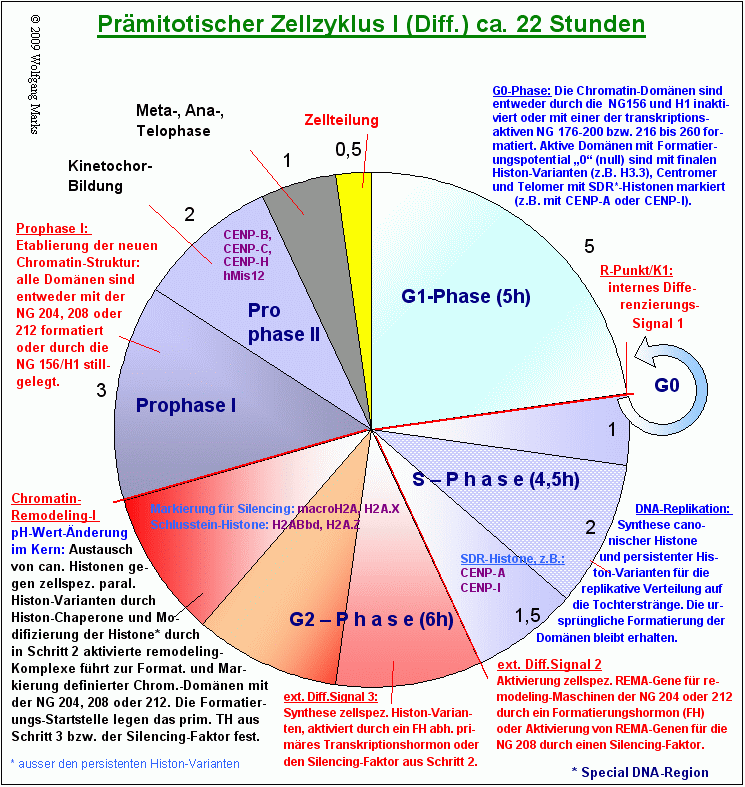
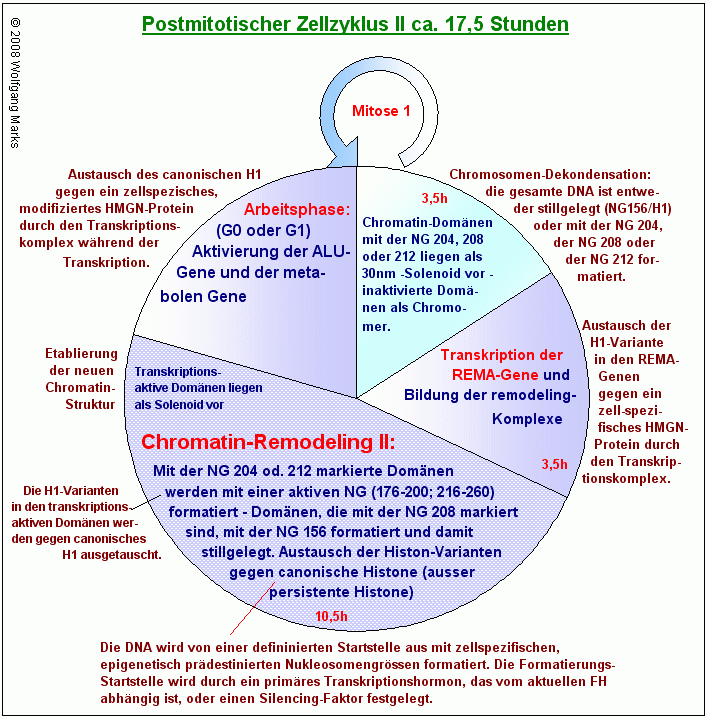
Da der Austausch von canonischen Histonen gegen paraloge Histon-Varianten postreplikativ, also entweder in der späten S-Phase oder in der G2-Phase stattfindet , kann die Markierung von Chromatin-Domänen mit den Nukleosomengrössen 204 oder 208 oder 212 nur in einem zweiten postmitotischen Zellzyklus an die Tochterzellen weitergegeben werden. Ich habe deshalb normatives und institutionelles/ epigenetisches remodeling wie in den Grafiken Zellzyklus I und Zellzyklus II gezeigt - auf zwei Zellzyklen verteilt.
Die Markierung von Chromatin-Domänen mit den Nukleosomengrössen 204, 208 und 212 wird durch Histon-Varianten vermittelt.
Die epigenetische Markierung von Chromatin-Domänen mit den Markierungs- oder Sondergrössen 204, 208, 212 erfolgt in meinem Modell durch den Einbau spezifischer Histon-Varianten während des prämitotischen Zellzyklus, der von ATP-abhängigen remodeling-Maschinen und Histon-Chaperonen katalysiert bzw. unterstützt wird. Die replikationsunabhängige Synthese dieser Markierungs-Histone ist von einem primären Transkriptionshormon (204, 212) oder einem Silencing-Faktor (208) abhängig. Während das primäre Transkriptionshormon von einem übergeordneten Formatierungshormon aktiviert wird (Beispiel: TRH -> TSH), das einen Teil der Formatierungskaskade darstellt, wird der Silencing-Befehl wahrscheinlich durch ein spezifisches Silencing-Protein der SATB-Familie vermittelt, das anstelle eines primären Transkriptionshormons zunächst in den Transkriptionskomplex (REMA-Gene und Gene für die Histon-Varianten) und anschließend in den Aktivierungskomplex (REMA-Gene) (Abb.) eingebaut wird. Dieser zellspezifische Silencing-Faktor bestimmt analog zu den Verhältnissen bei der Formatierung mit transkriptionsaktiven Nukleosomengrössen auch die Startstelle für die Formatierung der Domäne. Erst nach der Zellteilung, der Dekondensation des Chromatins und der Aktivierung der REMA-Gene werden im postmitotischen Zellzyklus II (Abb.) die solcherart markierten Chromatin-Domänen wiederum durch ATP-abhängige remodeling-Komplexe136 endgültig mit den epigenetisch definierten Nukleosomengrössen formatiert.
Da die Zahl der entdeckten Histon-Varianten ständig steigt und die Zahl der dazu veröffentlichen Arbeiten im gleichen Maße zunimmt, ist es im Rahmen dieser Arbeit nicht möglich und wohl auch nicht sinnvoll, alle bis dato bekannten Varianten und die ihnen zugeschriebenen Funktionen und Eigenschaften zu repetieren und zu referieren. Ich habe mich deshalb auf einige wenige Varianten beschränkt, von denen relativ gesicherte Erkenntnisse vorliegen und versucht, in der beiden Grafik Zellzyklus I diese Histon-Varianten mit bestimmten Phasen der Mitose zu verknüpfen. Dabei habe ich die Erkenntnisse berücksichtigt, die meine Voruntersucher zu diesen Varianten erarbeitet und zusammengetragen haben, soweit sie mir bekannt wurden.
Ein logischer, aus meinem Modell der Formatierten DNA direkt herleitbarer Ablauf der beiden Zellzyklen anlässlich einer differentiellen Zellteilung wäre demnach der folgende:
|
|
|
Der differentielle Zellzyklus ein Zwei-Zellzyklen-Modell.
Das Histon-Oktamer und die DNA eine mehr oder weniger fesselnde Beziehung.
Das Histon-Oktamer besitzt abhängig von den physiologischen Bedingungen eine a priori "zweigeteilte" oder auch viergeteilte Struktur, die das splitting bzw. das Deassemblieren der Tetramere und Dimere bei remodeling-Prozessen und während der Replikation der DNA abhängig von spezifischen HistonModifikationen in definierten Ebenen und Einheiten prädestiniert.
Bei in vitro-Versuchen mit DNA- und Oktamer-Lösungen bleiben die Histon-Oktamere in hohen Salzkonzentrationen stabil; bei Erniedrigung der Salzkonzentration jedoch zerfallen sie in ihre Bestandteile. Die Interaktionen zwischen den einzelnen Dimeren und Tetrameren selbst scheinen jedoch auch bei relativ niedrigen Salzkonzentrationen in vitro stabil zu sein.
Wird in vitro zu einer Oktamerlösung bei hoher Salzkonzentration DNA hinzugefügt und anschließend die Salzkonzentration langsam verringert, bindet die DNA an definierten Positionen an das Oktamer. Diese Kontakte zwischen DNA und Histon-Proteinen hielt man bis vor kurzem noch für unspezifisch (d.h. von der DNA-Sequenz unabhängig) - sie beruhen auf ionischen Wechselwirkungen, Wasserstoffbrückenbindungen und hydrophoben Wechselwirkungen zwischen dem DNA-Rückgrat und dem Histon-Oktamer. Bringt man assemblierte Nukleosom-Core-Partikel wieder in einen Puffer mit erhöhter Ionenstärke, so dissoziiert die DNA von den Komplexen und die Oktamere deassemblieren erneut und zerfallen dabei in ein (H3-H4)2 Tetramer und zwei H2A-H2B-Dimere. Assemblierung und Deassemblierung eines Corekomplexes aus einem Histon-Oktamer und DNA sind also in Abhängigkeit von der Ionenstärke reversibel.
Da die oben beschriebenen Versuche in vitro eine relativ große Stabilität des (H3-H4)2 Tetramers zeigten, glaubte man bis vor kurzem, dass die Histon-Oktamere beim Durchgang der Replikationsgabel während der DNA-Replikation in ein (H3-H4)2 Tetramer und zwei (H2A-H2B) -Dimere zerlegt und so auch auf die Tochterstränge verteilt werden. Das würde aber bedeuten, dass bei zufälliger, paritätischer Verteilung nicht jedes neue Nukleosom der beiden Tochterstränge ein parentales (H3-H4)2 Tetramer erhalten könnte, wie es bei der Verteilung der (H2A-H2B) -Dimeren auf die Tochterstränge der Fall ist. Bei der ungleichmässigen Verteilung der (H3-H4)2 Tetramere könnte dieses Tetramer also auch nicht wie ein H2A-H2B-Dimer - als template, also als Vorlage für die Vervollständigung oder das Auffüllen des aktuellen Tetramers/Oktamers dienen es müsste also nach einem speziellen Mechanismus für die Assemblierung der Oktamere während der Replikation gesucht werden.137
Eine Lösung dieses Problems scheinen neuere Untersuchungen zu bieten: wie wir schon seit geraumer Zeit wissen, sind Histone, wenn sie nicht gebraucht werden, an Chaperone gebunden. Bis vor kurzem glaubte man, dass Chaperone keine oder nur geringe Substrat-Spezifität besitzen.
Eine Studie von Tagami et al. (2004)138, die sich mit Histon-Chaperonen, ihren Funktionen und Beziehungen zu bestimmten Histonen befasst, zeigt nun, dass Histon-Chaperone offensichtlich präferentiell an bestimmte Histone binden: das Chaperon CAF1 bindet bevorzugt das S-PhaseHiston H3, also ein canonisches Histon -, das Chaperon HIRA dagegen scheint spezifisch die Histon-Variante H3.3 zu binden. Dies stimmt überein mit Ergebnissen des Labors Almouzni und Nakatani (Institut Pasteur, Paris), die in einem Xenopus-in-vitro-System demonstrieren konnten, dass das Chaperon HIRA Histone replikationsunabhängig requiriert, CAF1 dagegen nur während der S-Phase, also während der DNA-Replikation, als Histon-Chaperon aktiv ist.
Das eigentlich Überraschende war, dass Tagami et al. zeigen konnten, dass diese beiden Komplexe immer nur eine einzelne Kopie des jeweiligen H3- bzw. H3.3 Histons und ein H4-Monomer binden. Dies ergibt aber ein H3-H4- oder H3.3-H4-Dimer und kein (H3-H4)2 Tetramer!
Dieser auf den ersten Blick kleine Unterschied hat auf die Theorie der replikativen Verteilung der Histone in der S-Phase große Auswirkungen: wenn sich die Entdeckungen von Tagami et al. bestätigen (woran ich keinen Zweifel hege), wäre für H3-H4-Dimere (oder heterogene Dimere aus einem canonischen und einem varianten Histon) der gleiche Verteilungs-Mechanismus gegeben wie für solche von H2A-H2B. Die jeweils parentalen Dimere könnten somit bei der Replikation der DNA als Vorlage für die Vervollständigung der Oktamere der beiden Tochterstränge dienen und dabei die vorhandenen epigenetischen Markierungen/Informationen weitergeben.
Der Prämitotische Zellzyklus.
Drei Differenzierungssignale steuern das Chromatin-Remodeling.
In der Interphase des Zellzyklus sind die Chromatin-Domänen des gesamten Genoms entweder mit der NG150 formatiert und mit H1 assoziiert139 und damit stillgelegt oder mit einer der transkriptionsaktiven Nukleosomengrössen 176 bis 200 oder 216 bis 260 formatiert. Da eine bestimmte Chromatin-Domäne im Verlauf der Ontogenese nicht mit unendlich vielen, sondern nur mit einer epigenetisch festgelegten Zahl von Nukleosomengrössen formatiert werden kann, wird es im Leben jeder Zelle für eine bestimmte Chromatin-Domäne einen Zeitpunkt geben, wo das Formatierungspotential dieser Domäne erschöpft, der endgültige Formatierungs- und damit Differenzierungsstatus dieser spezifischen Domäne also erreicht ist. Dies kann nach zwei, vier, fünf, zwölf oder auch erst nach sechzehn verschiedenen (transkriptionsaktiven) Formatierungen der Fall sein. Die Nukleosomen solcher Domänen mit Formatierungspotential null werden in meinem Modell mit spezifischen finalen oder Schlußstein-Histonen wie zum Beispiel dem Histon H2ABbd oder H3.3 (replikationsunabhängig und persistent) gekennzeichnet und die Domäne dadurch (und durch andere, an SMARs gekoppelte Mechanismen) von weiteren Formatierungen ausgenommen.
Noch zwei weitere bekannte Histon-Varianten werden wir wahrscheinlich im Chromatin der Interphase finden: das bereits als H1-Variante bekannte CENP-A und die Variante CENP-I, die nach meiner festen Überzeugung ebenfalls eine H1-Variante darstellen. CENP-I und CENP-A gehören zu den Proteinen, die bis jetzt ausschließlich im Chromatin der Zentromer-Region gefunden wurden. Sie sind eng mit der Bildung des Kinetochors verknüpft. Ein deutsche Forschergruppe um S. Diekmann und P. Hemmerich vom Fritz-Lipmann-Institut in Jena haben 2008 berichtet140, dass von allen in Echtzeit mittels Fluoreszenz-Markierung in lebenden Zellen beobachteten Proteinen nur CENP-I und CENP-A während der Mitose im Zentromer gebunden bleiben. Die Bindung der anderen beobachteten Proteine (CENP-B, CENP-C, CENP-H und hMis12) an das Zentromer dagegen hat sich während des Zellzyklus drastisch verändert: es fand ein ständiger Austausch statt.
Spezielle DNA-Regionen werden durch spezifische Histon-Varianten markiert.
Diese Beobachtungen stützen meine These, dass auch CENP-I eine zentromer-spezifische H1-Variante ist.141 Beide Proteine CENP-A ebenso wie CENP-I - werden während der Replikation der DNA wahrscheinlich in der späten S-Phase - synthetisiert und in die Nukleosomen von Zentromer und vermutlich auch Telomer eingebaut. Ich habe diese Histon-Varianten, weil sie nur in speziellen DNA-Regionen vorkommen, deshalb SDR-Histone genannt142.
In der G1-Phase enthalten die Nukleosome weit überwiegend (zu etwa 90 %) canonische Histone die restlichen 10 % bilden sowohl replikationsabhängige (z.B. CENP-A, CENP-I) als auch replikationsunabhängige Histon-Varianten (z.B. H3.3, mH2A, H2A.Z). Allen diesen Histonen ist gemeinsam, dass sie mit spezifischen Funktionen verknüpft sind und einmal in die Nukleosomen eingebaut, als permanente Komponente in diesen verbleiben. Zusammenfassend werde ich diese Histone von nun an als persistente Histon-Varianten bezeichnen.
Beim Übertritt von der G0- oder der Interphase in die G1-Phase werden in der differenzierungswilligen Zelle zunächst die Proteine und Faktoren synthetisiert, die für die anstehende Replikation der DNA benötigt werden. Nach positiv verlaufenem Gesundheitscheck am R- bzw. K1-Punkt tritt die Zelle in das S-Stadium ein und repliziert die DNA. Die Frage, ob die Histone in Form von zwei H2A-H2B- und zwei H3-H4-Dimeren oder als zwei H2A-H2B-Dimere und ein H3/H4 Tetramer (H3-H4)2 auf die Tochterstränge verteilt werden, scheint mir nach den zuvor zitierten Studien nicht mehr offen zu sein: nach der zwei Dimere ein Tetramer -Theorie wäre die Weitergabe epigenetischer Informationen bei der Replikation der DNA nicht, bei der vier-Dimere-Theorie dagegen sehr leicht möglich. Auch meine Berechnungen und Überlegungen zur epigenetischen Markierung von Chromatin-Domänen mit den NG 204, 208 oder 212 haben wie man weiter unten sehen wird zu dem Ergebnis geführt, dass diese Markierung durch den Austausch von definierten canonischen Histon-Dimeren oder Monomeren gegen paraloge Histon-Varianten erfolgt, da sonst die Weitergabe der Markierung/Information in Zellzyklus II nicht möglich wäre.
Es muss ja nicht nur die DNA, sondern auch die Chromatin-Struktur repliziert werden, und es müssen die Informationen kopiert werden, die durch das Chromatin kodiert sind. Dazu müssen die Oktamere während der Replikation aber in Dimere und Monomere zerlegt werden.
Genom-Bereiche, die nicht mehr verändert werden, werden mit persistenten Histon-Varianten markiert.
Während der DNA-Replikation im prämitotischen Zellzyklus werden also sowohl canonische Histone benötigt - und synthetisiert - als auch persistente Histon-Varianten wie CENP-A, CENP-I, H3.3, mH2A, H2A.Z und H2A.Bbd, die allesamt zur Markierung von unveränderbaren genomischen Regionen verwendet werden.
Eine Zelle, die aus der G1-Phase in die Differenzierungsphase eintreten will, benötigt in meinem Modell während des prämitotischen Zellzyklus zwei bzw. drei Differenzierungssignale:
ein internes (am oder im Zusammenhang mit dem R-Punkt (auch K1-Punkt genannt) -
ein externes zu Beginn der G2-Phase: ein Formatierungshormon-
und ein drittes (externes) Signal in Form eines primären Transkriptionshormons, das vom aktuellen Formatierungshormon abhängig ist.143
Das Differenzierungssignal zwei, das aktuelle Formatierungshormon aktiviert nach dem im Kapitel über die REMA-Gene beschriebenen Muster über die Bindung an zellspezifische, frei zugängliche ASP des aktuellen Chromosomenbandes die Transkription von REMA-Genen, die abhängig vom epigenetischen Programm der Zelle - Remodeling-Faktoren für die Nukleosomengrößen 204 (ändern), 208 (stillegen) oder 212 (kopieren) kodieren.
Das Differenzierungssignal drei das vom aktuellen Formatierungshormon aktivierte primäre Transkriptionshormon stösst im Zellkern die (replikationsunabhängige) Transkription und Translation von zellspezifischen Varianten der Histone H1, H2A, H2B und H3 an, die anschliessend von den zuvor aktivierten remodeling-Maschinen (in die auch Chaperone, Ligasen, Kinasen eingebunden sind) nach dem im folgenden beschrieben Muster gegen canonische Histone ausgetauscht und gleichzeitig an distinkten Aminosäureresten modifiziert werden.
In Zellen, in denen Gene stillgelegt werden sollen, aktiviert das primäre Transkriptionshormon zusätzlich die Transkription eines spezifischen Silencing-Proteins.
Zur Erinnerung:
Chromatin-Domänen, deren Formatierung verändert werden soll, werden mit der Nukleosomengrösse 204 markiert (Domäne anders/neu formatieren).
Chromatin-Domänen, deren Formatierung gelöscht werden soll, werden mit
der Nukleosomengrösse 208 markiert (Domäne stillegen/inaktivieren).
Chromatin-Domänen, deren Formatierung übernommen werden soll, werden
mit der Nukleosomengrösse 212 markiert (Format der Domäne kopieren)
Im nächsten Schritt werden die canonischen Histone unter der Mitwirkung von REMA-Gen-kodierten Histon-Chaperonen und ATP-abhängigen, durch REMA-Gene kodierten remodeling-Maschinen für die Nukleosomengrössen 204, 208 und 212 durch zellspezifische paraloge Histon-Varianten nach folgendem Muster ersetzt:
In Chromatin-Domänen, deren Nukleosomenkonfiguration/Nukleosomengrösse geändert werden soll, werden in den Histon-Oktameren ein canonisches H2A-H2B- und ein canonisches H3-Dimer durch Dimere aus zellspezifischen paralogen Histon-Varianten und das Standard-H1-Molekül durch eine stammzellspezifische H1-Variante ersetzt.145
In Chromatin-Domänen, die inaktiviert werden sollen, werden in den Histon-Oktameren ein canonisches H2B-Monomer und ein canonisches H3-Monomer durch Monomere aus zellspezifischen paralogen Histon-Varianten und das Standard-H1-Molekül durch eine stammzellspezifische H1-Variante ersetzt.
In Chromatin-Domänen, deren Nukleosomenkonfiguration/Nukleosomengrösse kopiert werden soll, wird in den Histon-Oktameren ein canonisches H3-Dimer durch ein H3-Dimer aus einer zellspezifischen paralogen Histon-Variante und das Standard-H1-Molekül durch eine stammzellspezifische H1-Variante ersetzt.
|
|
|
|
|
Die Abläufe während einer differentiellen Mitose unterscheiden sich wesentlich von denen während einer proliferativen Zellteilung. Es muss ja nicht nur die DNA repliziert werden, sondern auch die Chromatin-Konfiguration, die innere Organisation der Chromatin-Komponenten neu strukturiert werden. Dazu bedient sich die Zelle spezifischer Remodeling-Maschinen und spezifischer Histon-Varianten, die sie zur Markierung der geplanten Veränderungen einsetzt.
|
|
|
Zeitgleich mit dem Austausch der canonischen Histone gegen zellspezifische paraloge Varianten nach dem zuvor beschriebenen Muster werden die inkorporierten Histon-Varianten, das variante H1, die canonischen Histone und definierte Nukleotide der DNA durch die zuvor erwähnten spezifischen remodeling-Maschinen für die NG 204, 208 und 212 an definierten Positionen modifiziert und als Folge dessen die nun heterogenen Histon-Oktamere mit dem gebundenen varianten H1 auf der DNA derart positioniert, dass die Chromatin-Domänen der betreffenden Zelle anschliessend entweder mit der NG 204, der NG 208 oder der NG 212 formatiert sind.
Jede einzelne Chromatin-Domäne der Zelle ist, damit abhängig von ihrem epigenetischen Programm, für die weiteren Differenzierungsschritte während des postmitotischen Zellzyklus eindeutig markiert.
In diesem Zusammenhang stellt sich zwangsläufig die Frage, was mit den persistenten Histon-Varianten in dieser Phase des Zellzyklus geschieht. Nun, wahrscheinlich... nichts. Im Hinblick auf die spezifischen Eigenschaften und die damit verbundenen Funktionen dieser Proteine im Rahmen der Kinetochor-Bildung, des silencing und des Differenzierungsstopps würde ein Austausch sicher zu gravierenden zellulären Veränderungen und zur Apoptose führen. Gleiches gilt für die chemische Modifikation von Aminosäureresten dieser Histone. In meinem Modell bleiben die persistenten Histon-Varianten in dieser Phase des Zellzyklus deshalb unverändert.
Der Postmitotische Zellzyklus
Nach der Zellteilung ist die gesamte Kern-DNA der beiden Tochterzellen entweder durch die Nukleosomengrösse 150 in Verbindung mit spezifisch modifiziertem canonischem H1 inaktiviert oder mit der NG 204, mit der NG 208 oder der NG 212 formatiert und damit zugleich zellspezifisch für die folgende Formatierung mit einer transkriptionsaktiven Nukleosomengrösse oder für die Stillegung durch Formatierung mit der NG 150 markiert .
In allen Chromatin-Domänen mit Ausnahme der bereits inaktivierten Domänen ist das canonische H1-Histon wie zuvor beschrieben durch eine stammzellspezifische H1-Variante ersetzt worden.
|
|
|
|
|
Im postmitotischen Zellzyklus werden die markierten Chromatin-Domänen mit der epigenetisch programmierten (oder auch determinierten) Nukleosomengrösse neu formatiert. Die Markierungs-Histone (Histon-Varianten) werden mit Ausnahme der persistenten Histon-Varianten wieder gegen canonische Histone ausgetauscht.
|
|
|
Damit die DNA der Zelle in einem zweiten remodeling-Schritt mit den epigenetisch definierten Nukleosomengrössen formatiert werden kann, müssen die Chromosomen nach der Zellteilung zunächst wieder entpackt werden. Bei diesem Entpackungsvorgang spielen Architekturproteine, insbesondere solche der HMGA, SATB- und der SAF-Familie eine entscheidende Rolle.
Während Chromatin-Domänen, die mit einer der NG 204, 208 oder 212 formatiert sind, vermutlich bis zur 30nm-Struktur, dem Solenoid dekondensiert werden, werden bereits inaktivierte Chromatin-Domänen mit einiger Wahrscheinlichkeit als Chromomer-Struktur fixiert. Sie auszupacken würde auch wenig Sinn machen.
Damit die REMA-Gene transkribiert und translatiert werden können, muss das an die Oktamere gebundene linker-Histon, das bei den mit der NG 204, 208 und 212 formatierten Domänen aus einer formatierungshormon-spezifischen H1-Variante besteht, gegen einen aktivierten Komplex aus einem HMGN-Protein und einem Hormon-Rezeptorproteinkomplex ausgetauscht werden. Dies geschieht bei der Bildung der Transkriptionsschleife: die Bindung des HMGN/HRP-Komplexes führt über die Öffnung des Solenoids zu einer transkriptionsfähigen 10nm-Perlenketten-Struktur, der einzigen DNA-Struktur, in der die transkriptionsrelevanten DNA-Sequenzen für die Transkriptionsmaschine zugänglich sind.
Im Anschluss an die Installation der transkriptionsfähigen Chromatin-Struktur werden in den mit der NG 204, 208 und 212 formatierten Domänen unter dem Einfluss des aktuellen Formatierungshormons146 (NG 204, 212) bzw. unter dem Einfluss eines Silencing-Faktors (NG208) auf dem im Kapitel über die REMA-Gene detailliert beschriebenen Weg spezifische REMA-Gene aktiviert und Remodeling-Faktoren synthetisiert, die sich zu remodeling-Maschinen zusammenschließen und die mit 204, 208 oder 212 formatierten und markierten Domänen mit einer prädestinierten, epigenetisch programmierten Nukleosomengrösse formatieren. Dabei werden die H1-Varianten der dem remodeling unterworfenen Domänen und die als linker-Ersatz fungierenden HMGN/HRP-Komplexe in den REMA-Genen wieder gegen canonische H1-Histone ausgewechselt.147
Nach Vollendung des remodeling-Prozesses sind die zuvor mit der NG 208 (stillegen) markierten Domänen also mit der NG150 formatiert und in Verbindung mit einem spezifisch modifizierten H1-Histon permanent stillgelegt, Domänen mit der NG 212 (kopieren) mit der alten NG der Mutterzelle formatiert, Domänen mit der NG 208 (ändern) mit einer neuen NG formatiert, die dem aktivierenden, aktuellen Formatierungshormon entspricht.
Zur Erinnerung: die hier für die differentielle Mitose beschriebenen Remodelingprozesse sind auf der Ebene von Chromosomenbändern organisiert.
Die Neuformatierung einer Chromatin-Domäne mit einer transkriptions-
aktiven Nukleosomengrösse im prämitotischen Zellzyklus.
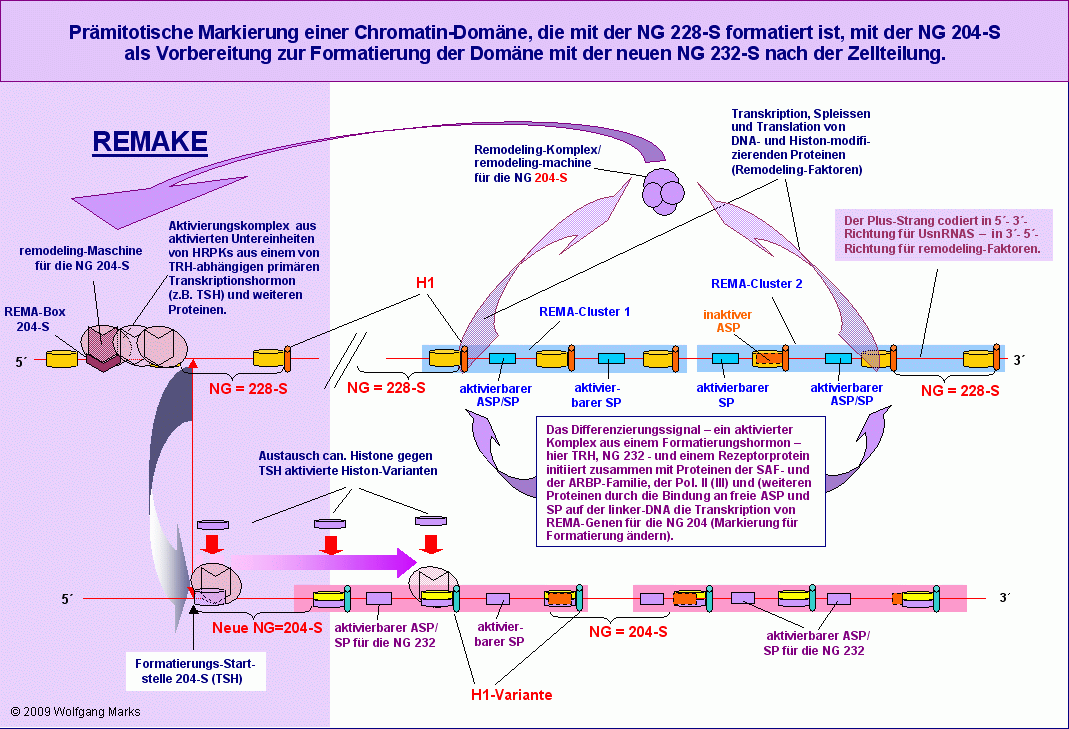
Die Formatierung einer aktiven Chromatin-Domäne148 während einer differentiellen Zellteilung verläuft also in zwei Phasen. Im prämitotischen Zellzyklus wird die betreffende Domäne durch eine der Nukleosomengrössen 204 oder 208 oder 212 markiert im postmitotischen Zellzyklus entweder mit einer neuen Nukleosomengrösse formatiert, durch Formatierung mit der NG 150 stillgelegt oder wieder mit der alten Grösse formatiert. Um die hier beschriebenen und schematisch in den Grafiken Zellzyklus I und II dargestellten Prozesse verständlicher zu machen, habe ich die Abläufe während der beiden Zellzyklen der Mitose in je einer Grafik versucht weiter zu verdeutlichen. Ich bin davon ausgegangen, dass die dargestellte Domäne mit der NG 228 formatiert ist und mit der epigenetisch programmierten NG 232 formatiert werden soll. Zum besseren Verständnis und zum Vergleich sollten die Grafiken Zellzyklus I und Zellzyklus II herangezogen werden.
Die Neuformatierung wird initiiert durch Bindung eines spezifischen Formatierungshormons (im gezeigten Beispiel TRH) an einen zellulären (kario- oder zytoplasmatischen) Rezeptor, der danach in aktive Untereinheiten zerfällt. Diese Untereinheiten binden zusammen mit Proteinen der SAF- und der ARBP-Gruppe sowie anderen Proteinen als Aktivierungskomplex an ein Netzwerk von REMA- und UsnRNA-Genen für die NG 204, das durch die aktuelle Formatierung der Domäne mit der NG 228 definiert wurde.
Wie im Kapitel über die REMA- und die UsnRNA-Gene beschrieben, werden zunächst die UsnRNA-, danach die REMA-Gene transkribiert. DieTranskripte der REMA-Gene werden wie bei einem normalen metabolischen Gen mit Hilfe der zuvor generierten UsnRNAs gespleisst, die gespleisste messenger-RNAs ins Zytoplasma transportiert und dort translatiert und gegebenenfalls weiterverarbeitet . Dies können spezifische Modifikationen sein, aber auch eine Aufspaltung des Polypeptids in zwei (oder mehr) verschiedene Remodeling-Faktoren.
Die fertigen Proteine werden zurück in den Zellkern transportiert und schließen sich dort mit den anderen Proteinen des Netzwerks dynamisch zu einer remodeling-Maschine zusammen. Diese bindet durch Vermittlung eines Aktivierungskomplexes149, der unter anderem HRPKs aus einem vom aktuellen Formatierungshormon abhängigen primären Transkriptionshormon enthält, im REMAKE der Domäne an eine spezifische REMA-Box für die NG 204.
Es ist auch das primäre Transkriptionshormon, das zu Beginn der G2-Phase die Synthese von Histon-Varianten anstösst, die im weiteren Verlauf der G2-Phase durch die zuvor generierten remodeling-Maschinen in die Histon-Oktamere eingebaut und zellspezifisch so modifiziert werden, dass die Domäne anschliessend mit der Nukleosomengrösse 204 formatiert und markiert ist.
|
|
|
|
|
|
|
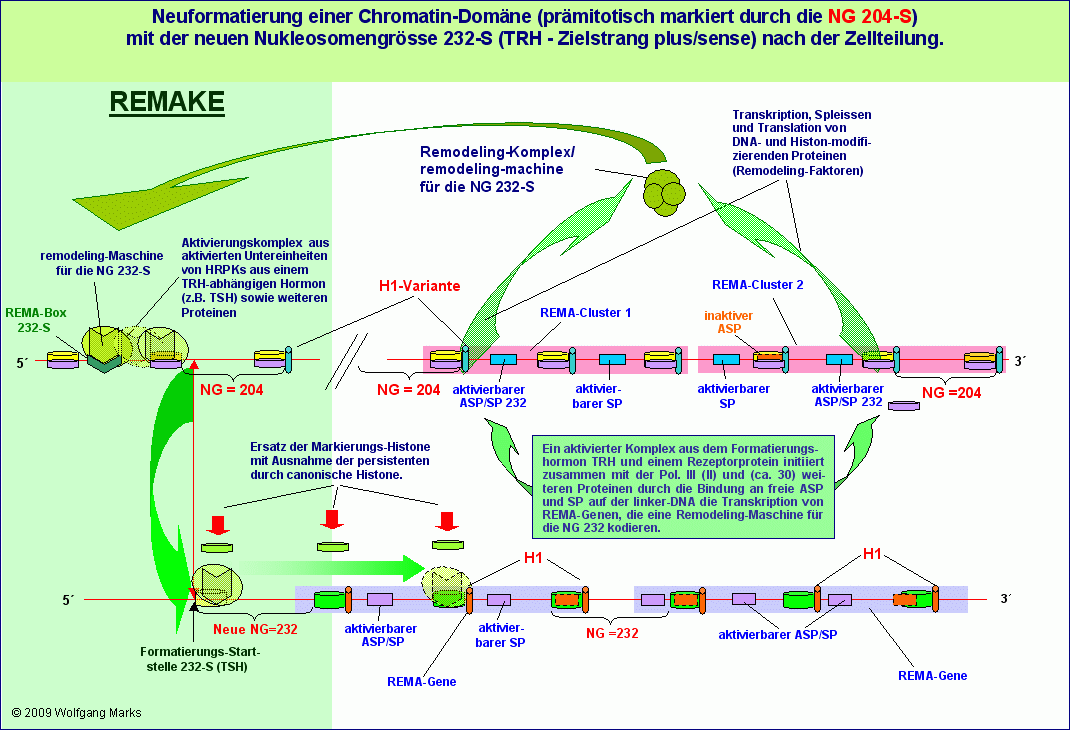
Die Neuformatierung einer Chromatin-Domäne mit einer transkriptionsaktiven Nukleosomengrösse im postmitotischen Zellzyklus
Nach der Zellteilung ist unsere Beispiel-Domäne also mit der Nukleosomengrösse 204 der Markierung für Nukleosomengrösse der Domäne ändern formatiert.
Die Formatierung unserer Domäne mit der NG 204 führt wie in der Abbildung gezeigt wiederum unter Beteiligung des aktuellen Formatierungshormons - zur Aktivierung von epigenetisch determinierten REMA-Genen für eine remodeling-Maschine für die Nukleosomengrösse 232, die nach dem bekannten Muster durch Vermittlung eines Aktivierungskomplexes und des primären Transkriptionshormons an die REMA-Box 232-S im REMAKE der Domäne bindet (Zielstrang der Formatierung ist der plus-Strang) und die Domäne von einem definierten Startpunkt aus, der durch das primäre Transkriptionshormon bestimmt ist, mit der NG 232-S formatiert.
Wie im Kapitel über die REMAKEs ausgeführt, kann die Formatierung einer Chromatin-Domäne mit einer bestimmten Nukleosomengrösse abhängig vom jeweils aktiven primären Transkriptionshormon in verschiedenen Zellen oder in verschiedenen Differenzierungsstufen der gleichen Zelle von verschiedenen Startpunkten aus erfolgen. Dieser Startpunkt, die Formatierungs-Startstelle (FSS) entscheidet letztlich über die Positionierung aller Nukleosomen in der konnektierten Chromatin-Domäne und damit auch über die Zugänglichkeit von Promotoren, Enhancern, poly-A-Signalen und anderen regulativen DNA-Sequenzen. Jeder dieser Startpunkte steht aber auch, wie ich in Teil III zeigen werde, in einer direkten arithmetischen Beziehung zu einer spezifischen Transkriptionsstartstelle (TSS), die demnach sowohl mit dem Formatierungshormon, als auch mit dem primären Transkriptionshormon korreliert ist.
Ob und wie ein polycistronisches Gen exprimiert wird, welche Exons eines bestimmten Gens transkribiert werden, hängt also explizit von der Formatierung des Gens (der Domäne) mit einer bestimmten Nukleosomengrösse die durch das Formatierungshormon definiert ist - und von der Formatierungsstartstelle ab, die von einem primären Transkriptionshormon bestimmt wird.
Die aktuell gebildete remodeling-Maschine ist also mit einem der 19 transkriptionsaktiven Formatierungshormone korreliert die Formatierungs-Startstelle die Position des ersten Histon-Oktamers - von einem primären Transkriptionshormon bestimmt, das vom aktuellen Formatierungshormon aktiviert wurde.
Das Genom als Datenbank: epigenetische Differenzierung
basiert auf Zellprogrammen, die remodeling-Befehle
für Chromatin-Domänen enthalten.
Jede Zelle des menschlichen Körpers besitzt ein inhärentes epigenetisches Programm, das sie im Verlauf der Ontogenese abarbeitet. Für die Beschreibung dieses Programms gibt es mehrere Möglichkeiten:
I.
Für eine definierte Chromatin-Domäne einer differenzierungsfähigen, zunächst nur proliferierenden und sich anschliessend differenzierenden Zelle könnte dieses epigenetische Programm zum Beispiel wie folgt beschrieben (oder geschrieben) werden:
180-212-180-212-180-204-184-204-200-204-216-212-216-212-216-
Das Format der Domäne wird zunächst zweimal kopiert, dann viermal nacheinander verändert und schließlich noch zweimal kopiert. Der Bindestrich zeigt an, dass eine weitere Formatierung der Domäne abhängig von Differenzierungssignalen möglich ist.
II.
Das epigenetische Programm dieser Domäne ist dem oben beschriebenen zunächst ähnlich, die Domäne wird allerdings in einem bestimmten Stadium der Ontogenese dauerhaft inaktiviert.
200-212-200-212-200-204-216-204-220-204-232-212-232-208-150~
Um die Gene dieser Domäne stillzulegen, wird die Domäne zunächst mit der NG 208 markiert - Nukleosomen dieser Domäne enthalten dann u.a. die Histon-Variante H2A.X und schließlich durch Formatierung mit der NG 150 in Kombination mit der Bindung von modifiziertem H1 stillgelegt. Das Tildezeichen bedeutet, dass die Formatierung 150 (stillegen) mit der Bindung von H1 verknüpft und deshalb irreversibel ist.
III.
Der Zell- oder Genomstatus einer ausdifferenzierten Zelle, die nicht mehr proliferiert (eine Nervenzelle zum Beispiel) ist dadurch gekennzeichnet, dass der eine Teil der Chromatin-Domänen mit der NG 150 formatiert und mit spezifisch modifiziertem H1 bestückt ist - die Gene in diesen Domänen sind demnach stillgelegt - der andere Teil der Domänen zwar jeweils mit einer der transkriptionsaktiven Nukleosomengrössen (176-200;216-260) formatiert ist, Differenzierung aber gleichwohl nicht mehr möglich ist, da das entscheidende Differenzierungssignal - ein neues Formatierungshormon und/oder der Rezeptor dafür fehlen. Zudem sind diese noch transkriptionsaktiven Domänen einer ausdifferenzierten, nicht mehr proliferierenden Zelle wahrscheinlich durch den Einbau spezifischer Schlußstein- (finale) Histone in die Oktamere - wie das variante H2A.Bbd oder H3.3150 - zusätzlich vor weiteren Formatierungen geschützt.
Das epigenetische Programm zweier beliebiger Domänen im Genom einer solchen Zelle könnte demnach wie folgt geschrieben sein:
Domäne 1 (stillgelegt)
200-212-200-212-200-204-220-204-232-212-232-212-232-208-150~
Domäne 2 (Formatierungsstop)
200-212-200-212-200-204-224-204-228-204-232-204-236-204-244!
Um die Gene in Domäne 1 stillzulegen, wird die Domäne zunächst mit der NG 208 markiert - Nukleosomen dieser Domäne enthalten die Histon-Variante H2A.X und schließlich durch Formatierung mit der NG150 in Kombination mit der Bindung von H1 stillgelegt.
Die Expression der Gene in Domäne 2 wird im letzten Stadium der Differenzierung durch Hormone gesteuert, deren Synthese und Sekretion vom Formatierungshormon MSH-RH (NG244) abhängig ist. Das Ausrufezeichen signalisiert, dass die Formatierungskapazität dieser Domäne erschöpft ist. Die Nukleosomen dieser Domäne können zum Beispiel die Schlußstein-Histone H2A.Bbd oder H2A.Z enthalten.
Wieviele remodeling-Maschinen gibt es (oder muss es geben)?
Nach allem bisher Vorgetragenen dürfte offensichtlich sein, dass es eine zwar große, aber dennoch definierte und überschaubare Zahl von remodeling-Maschinen geben muss, die in den vermutlich 240151 verschiedenen Zellen, die aus der Zygote im Verlauf der Ontogenese entstehen, die epigenetisch prädestinierten Genomformate etablieren, die dem jeweiligen Entwicklungsstadium der Zelle entsprechen.
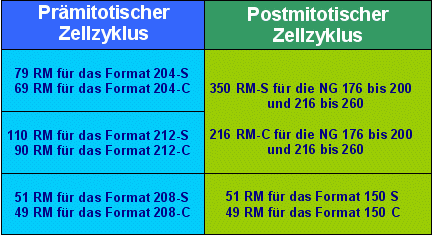 |
Auf der Basis der bis heute bekannten Histon-Modifikationen und meinen Hochrechnungen zu den möglichen Modifikationen an den Histonen, den Histon-Varianten und der DNA habe ich versucht, die Zahl der benötigten remodeling-Maschinen zu berechnen, weniger um eine genaue Zahl zu bekommen, sondern vielmehr um die Grössenordnung abzuschätzen. Obwohl die Analyse mehrerer REMAKEs mir gezeigt hatte, dass eine einzelne Chromatin-Domäne im Verlauf der Ontogenese mit bis zu 23 verschiedenen Nukleosomengrössen formatiert werden kann - neunzehn transkriptionsaktive Grössen pro DNA-Strang plus die NG 150, 204, 208 und 212 - hat das Ergebnis meiner Berechnungen zunächst auch mich überrascht: denn nach diesem Ergebnis würden 1114 verschiedene remodeling-Maschinen (RM) benötigt, um alle epigenetischen Muster, die im menschlichen Genom zur Zeit programmiert sind, während der Ontogenese zu realisieren. Allein für die Nukleosomengrösse 260 muss es insgesamt 26 verschiedene RM geben, nämlich 12 für die NG 260-C und 14 für die NG 260-S.
|
|
|
132Hier und im folgenden sind ATP-abhängige remodeling-machines gemeint.
133Wahrscheinlich gibt es für die Stillegung von Genen zwei oder drei verschiedene Mechanismen abhängig davon, ob eine ganze Chromatin-Domäne stillgelegt werden soll (=30nm-Struktur), oder nur eine einzelne Chromatin-Subdomäne (=10nm aggregierte Nukleosomenkette mit oder ohne H1.
134Genomformat oder Genomstatus die Gesamtheit aller Formatierungen der Chromatin-Domänen des Genoms.
136siehe Kapitel: Das REMA-Netzwerk
137Ein solcher wurde meines Wissens bis heute nicht gefunden.
138Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis; Tagami H, Ray-Gallet D, Almouzni G, Nakatani Y; Cell. 2004 Jan 9;116(1):51-61.
139Abhängig davon, ob eine einzelne Chromatin-Domäne oder das ganze Chromomer inaktiviert ist, liegt das Chromatin in unterschiedlich kondensierten Konformationen vor.
140Hemmerich P, Weidtkamp-Peters S, Hoischen C, Schmiedeberg L, Erliandri I, Diekmann S. Dynamics of inner kinetochore assembly and maintenance in living cells.(2008) J. Cell Biol. 180:1101-1114
141Vermutlich wird nicht nur das Zentromer, sondern auch das Telomer als zweite DNA-Region mit einer extrem wichtigen Funktion sowohl für die Chromatin-Kondensation zum Chromosom, als auch für das Überleben der Zelle mit spezifischen Histon-Varianten markiert.
142Special-DNA-Regions
143Beispiel: Formatierungshormon TRH, prim. Transkriptionshormon TSH. Die Hormone binden nach dem bekannten Muster an Rezeptoren in der Zellmembran oder der Kernmembran.
145Zur Erinnerung: ich postuliere 19 H1-Varianten.
146Siehe dazu auch die Bemerkungen im Abschnitt: Stillegung von Genen in einer Chromatin-Domäne durch Formatierung mit der Nukleosomengrösse 208.
147Der Austausch von H1 gegen einen HMGN-HRP-Komplex muss nach meinen Analysen ein allgemeines Phänomen bei der Transkription von Genen sein. Ich komme darauf später noch zurück.
148mit einer der transkriptionsaktiven Nukleosomengrössen 176 bis 200 und 216 bis 260...
149Der Aktivierungskomplex geht wahrscheinlich aus dem Transkriptions/Spleisskomplex hervor enthält aber deutlich weniger Proteine.
150Es sollte in meinem Modell eine definierte Anzahl solcher Schlußstein-Varianten von jedem Histon-Protein (ausser von H4) geben, die in ein Oktamer eingebaut - das Ende der Formatierungskaskade für die jeweilige Chromatin-Domäne markieren. Von den 84 postulierten Histon-Varianten gehören vermutlich 30 zu den Schlußstein-Histonen (SSH), im Einzelnen: H1=5 SSH; H2A=5 SSH; H2B=10 SSH; H3=10 SSH.
151Rechnet man die während einer Schwangerschaft gebildeten Zellen im Körper der Mutter hinzu, kommt man auf die Zahl 260, die ich eingangs dieser Arbeit genannt habe. Im adulten Organismus sind davon noch etwa 110 vorhanden.
|
|
|