|
|
 |
|
|
|
Wolfgang Marks: Die Formatierte DNA
|
|
|
Struktur und Organisation des Chromatins.
Chromosomenbänder, Transkriptionsgruppen, Domänen, Subdomänen.
Dass die DNA eine innere Struktur, eine inhärente Ordnung hat, geht schon aus der Tatsache hervor, dass sie während der Mitose nicht als mehr oder weniger zusammengeknäueltes Riesenmolekül an die Tochterzellen weitergegeben wird, sondern säuberlich verpackt in Form von 46 Chromosomen. Auch die Bänderung der Chromosomen ist ein Hinweis darauf, dass es unterhalb der Chromosomen-Ebene weitere Strukturen geben muss, welche die DNA in funktionelle Bereiche gliedern.
|
|
|
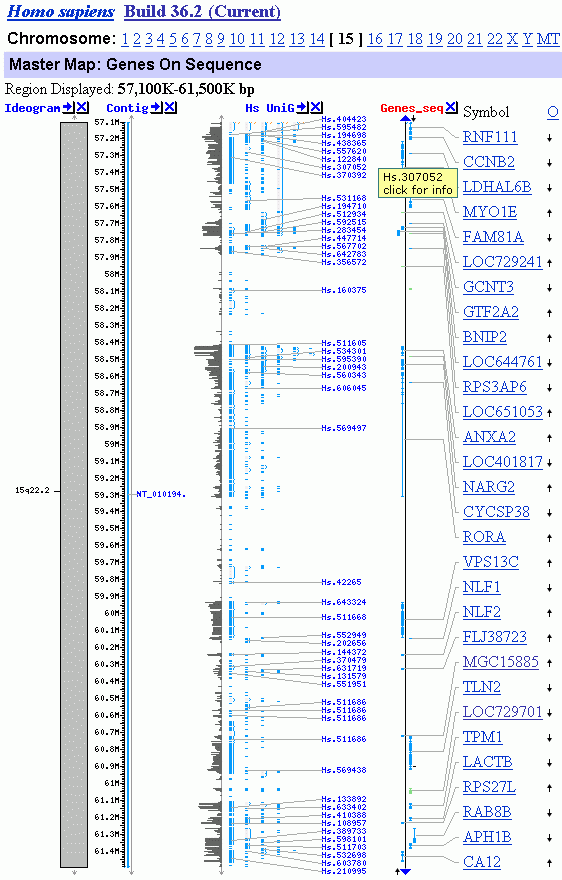
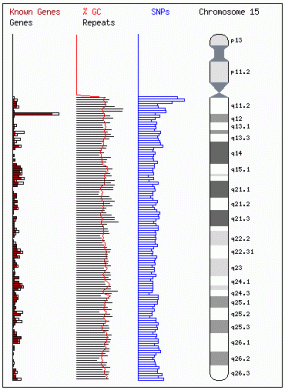 Die Abbildungen (Quelle: NCBI) zeigen das Chromosom 15 und einen Teil seines langen Arms mit dem Chromosomenband q22.2. In diesem Band liegt das TPM1-Gen (Tropomyosin-alpha), das ich ins Zentrum meiner Untersuchungen gestellt habe. Die Abbildungen (Quelle: NCBI) zeigen das Chromosom 15 und einen Teil seines langen Arms mit dem Chromosomenband q22.2. In diesem Band liegt das TPM1-Gen (Tropomyosin-alpha), das ich ins Zentrum meiner Untersuchungen gestellt habe.
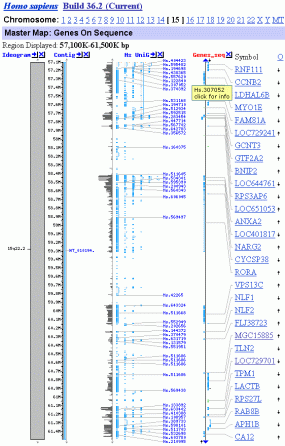 Region Displayed: 15q22.2 Region Displayed: 15q22.2
Region Displayed: 57100K-61500K bp
Total localized Genes On Chromosome: 1143
Genes Labeled: 30
Total Genes in Region: 42
|
|
|
Chromosomenbanden sind die sichtbaren Zeichen einer inneren Ordnung der DNA und können in bestimmten Stadien des Zellzyklus (z.B. in der Metaphase) durch verschiedene Färbe- und Extraktionsmethoden auf verschiedene Weise sichtbar gemacht werden. Wodurch die charakteristische Bänderung der Chromosomen entsteht, ist bis heute nicht bekannt. Auch über die Organisation der DNA in funktionelle Bereiche unterhalb der Ebene der Chromosomenbänder weiss man wenig. Ob es zwischen den Chromosomenbändern und den postulierten und in der Literatur immer wieder apostrophierten (aber bis heute in ihren Grenzen nicht definierten) Chromatin-Domänen weitere Strukturen gibt, die mit der Funktion der DNA und der Gene darin korreliert sind, ist nicht gewusst. Von einem kompletten Verständnis der zweifellos vorhandenen inneren Struktur der DNA und der Auswirkung dieser Struktur auf die Genexpression oder auf die Kompaktierung des Chromatins zum Chromosom und vice versa ist man noch weit entfernt.
|
|
|
|
|
Formatierung findet in den Grenzen von Chromatin-Domänen statt. Chromatin-Domänen lassen sich zu Transkriptionsgruppen
zusammenfassen Transkriptionsgruppen zu Chromosomenbändern.
Nach dem Ergebnis meiner Computer-Analysen mittels IMPACD® lässt sich ein Chromosomenband in abgrenzbare Transkriptionsgruppen, diese in Chromatin-Domänen und diese wiederum in Chromatin-Subdomänen einteilen. Jede dieser Einheiten weist spezifische Merkmale auf:
Ein Chromosomenband
umfasst mehrere Transkriptionsgruppen. Es stellt einen DNA-Abschnitt dar, in dem eine Gruppe stammzellspezifischer remodeling-Maschinen kodiert wird. Alle Chromatin-Domänen eines definierten Chromosomenbandes werden ausschliesslich durch remodeling-Maschinen formatiert, die auch in diesem Band kodiert werden.
Eine Transkriptionsgruppe
besteht aus mehreren Chromatin-Domänen. Sie repräsentiert eine Imprinting-Einheit. Alle Gene einer Transkriptionsgruppe werden vom selben DNA-Strang transkribiert.
Eine Chromatin-Domäne
bildet eine formatting unit - einen DNA-Abschnitt, der in einer definierten Zelle in allen Phasen des Zellzyklus eine einheitliche, distinkte Nukleosomengrösse aufweist.
Ein Chromatin-Subdomäne
definiert eine genetische Funktionseinheit; sie enthält alle Promotoren, Terminatoren und regulativen Gene, welche für die Generierung von spezifischen Primärtranskripten und messenger-RNAs der Gene in der Subdomäne erforderlich sind.
|
|
|
|
|
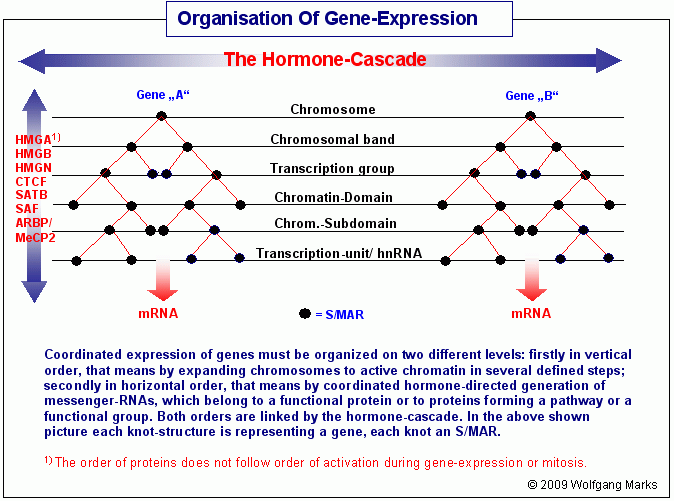
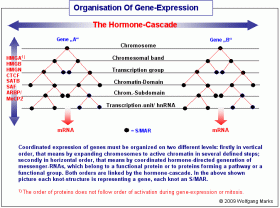
Koordinierte Genexpression erfordert eine vertikale und eine
horizontale Organisation der DNA.
Viele Proteine des menschlichen Organismus werden aus Proteindomänen zusammengefügt, die nicht von einem Gen, sondern von zwei (oder mehr wahrscheinlich bis zu vier ) Genen auf unterschiedlichen Genloci und Chromosomen kodiert werden. Dazu kommt, dass fast alle Proteine, welche die Zelle herstellt, zu bestimmten Enzymsystemen (pathways) oder funktionellen Gruppen gehören, ihre Synthese also eine genaue zeitliche (und räumliche) Koordination erfordert. Daher muss es ein umfassendes und vernetztes System geben, das sowohl die kontrollierte Dekondensation des Chromatins das ist die vertikale Organisation - als auch die zeitgleiche Aktivierung der Gene in den dekondensierten Bereichen und die einzelnen Stufen der Transkription und das Processing der Transkripte kontrolliert.104Letzteres nenne ich die horizontale Organisation.
.
 Wie wir aus dem zuvor Gesagten wissen, ist das Genom jeder einzelnen Zelle von der ersten Teilung der Zygote an einem beständigen Umbau unterworfen. Definierte Abschnitte der Zell-DNA werden bei jedem Differenzierungsschritt mit neuen, epigenetisch prädestinierten Nukleosomengrößen formatiert oder es wird die Nukleosomenkonfiguration solcher Abschnitte kopiert oder der ganze Abschnitt wird stillgelegt. Die REMA-Gene, welche die Faktoren kodieren, die sich dynamisch zu remodeling-Maschinen zusammenschließen, sind, wie ich weiter oben im Detail erklärt habe, auf jedem DNA-Strang durch ein Netzwerk von Promotor-Adressen zu Transkriptionseinheiten zusammengefasst. Wie wir aus dem zuvor Gesagten wissen, ist das Genom jeder einzelnen Zelle von der ersten Teilung der Zygote an einem beständigen Umbau unterworfen. Definierte Abschnitte der Zell-DNA werden bei jedem Differenzierungsschritt mit neuen, epigenetisch prädestinierten Nukleosomengrößen formatiert oder es wird die Nukleosomenkonfiguration solcher Abschnitte kopiert oder der ganze Abschnitt wird stillgelegt. Die REMA-Gene, welche die Faktoren kodieren, die sich dynamisch zu remodeling-Maschinen zusammenschließen, sind, wie ich weiter oben im Detail erklärt habe, auf jedem DNA-Strang durch ein Netzwerk von Promotor-Adressen zu Transkriptionseinheiten zusammengefasst.
Da den physikalischen Adressraum dieser beiden REMA-Netzwerke jeweils ein Chromosomenband bildet, formatieren die derart generierten remodeling-Maschinen für eine bestimmte Nukleosomengrösse deshalb auch chromosomenband-spezifisch das heißt, eine remodeling-Maschine für die NG 216 zum Beispiel, die im Chromosomenband 15q22.2 kodiert ist, müsste nicht notgedrungen auch im benachbarten Chromosomenband 15.q22.31 funktionieren.105
Obwohl alle Chromatin-Domänen eines Chromosomenbandes in einer gegebenen Zelle eine andere Formatierung aufweisen können (nicht müssen), richten sich die Modifikationen an den Histonen und der DNA, auf die Chromosomenbande bezogen, doch nach einem gemeinsamen System. Allerdings sind es nicht die typischen oder spezifischen Besonderheiten dieser Modifikationen innerhalb eines Chromosomenbandes, welche die dokumentierte Bänderung der Chromosomen verursachen, sondern es ist die Reduzierung dieses Systems auf einige wenige grundlegende Veränderungen an der DNA und den Histonen während der Mitose, die in der Metaphase sichtbar wird. Das System, das sich in der Bänderung der Chromosomen zeigt, ist ja nicht die Fotografie, die Abbildung des Arbeitszustandes der Zelle, sondern die des Transformations- oder Verpackungszustands ihres Chromatins in einer spezifischen Phase des Zellzyklus eben der Metaphase. In dieser Phase aber beschränkt sich die Formatierung der Chromatin-Domänen auf einige wenige charakteristische Modifikationen und Nukleosomengrössen. Dies werde ich anschließend im Kapitel über das chromatin-remodeling und die differentielle Mitose noch im Detail besprechen.
Mit wieviel verschiedenen Nukleosomengrössen eine einzelne Chromosomenbande bzw. die in ihr konfigurierten Domänen formatiert werden können, hängt, wie wir jetzt wissen, von Art und Bestand der REMA-Gene in diesem Band ab. In einem Chromosomenband werden aber mit einiger Wahrscheinlichkeit nicht alle möglichen Nukleosomengrössen genutzt, ebensowenig wie im ganzen Chromosom. Wahrscheinlicher ist, dass jedes Chromosom, jedes Band, jede Domäne ein definiertes Formatierungspotential hat: dabei muss dieses Potential in einem kleinen Chromosom wie dem Chromosom 22 nicht unbedingt geringer sein als in einem großen wie dem Chromosom 1 das Potential hängt wohl eher von der Zahl und Art der Gene ab, als von der absoluten Größe des Chromosoms in Basenpaaren gemessen.
Das Chromosom 22 zum Beispiel enthält (Stand 06/2008) 508 Strukturgene106 und zählt 49.691.432 Basenpaare. Das Chromosom 1 ist etwa 5-mal so groß, müsste also etwa 2500 Gene enthalten, wenn es eine direkte Relation zwischen der Zahl der Basenpaare und der Zahl der Strukturgene gäbe. Tatsächlich sind es aber ca. 20% weniger, nämlich nur 2153.
Das humane Genom ist hierarchisch organisiert.
Diese Organisation wird von Architekturproteinen und SMARS
übernommen.
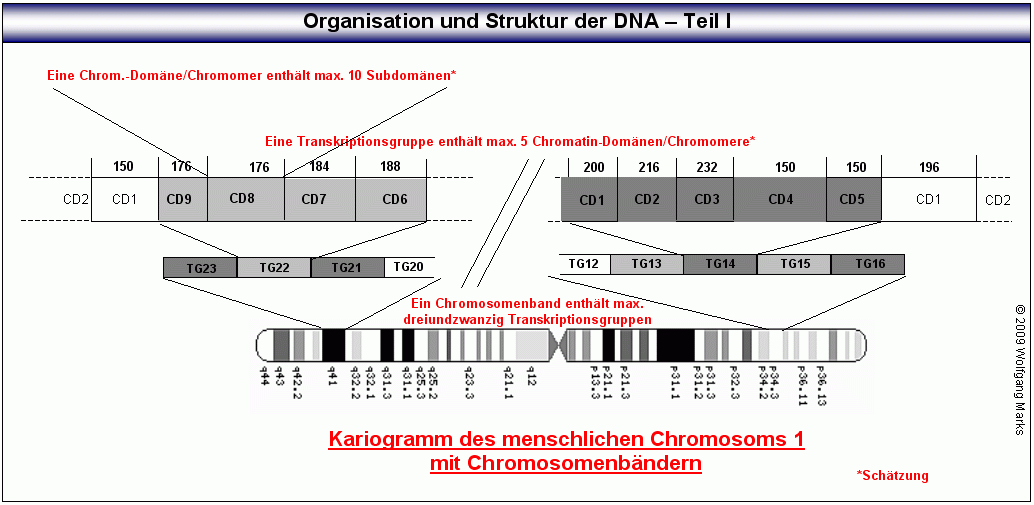
Es scheint logisch, dass ein Chromosomenband aus maximal 23 Transkriptionsgruppen (gleich der Zahl der verfügbaren Nukleosomengrössen) bestehen kann. Dass eine Transkriptionsgruppe maximal 5 Chromatin-Domänen/Chromomere zählen kann, ist eine Schätzung, die sich an der durchschnittlichen Grösse der Transkriptionsgruppen und der Domänen orientiert. Chromatin-Domänen wiederum und das ist eine Annahme, die sich an der Zahl der Differenzierungsstufen orientiert, die eine Zelle vermutlich höchstens durchlaufen kann - können wahrscheinlich bis zu 10 Subdomänen enthalten. Um die Hierarchie dieser Strukturen und damit ihre Funktionalität während der Kondensation und Dekondensation des Chromatins im Verlauf einer Mitose zu erhalten, muss es zweifellos Sequenzen geben, die nicht nur diese DNA-Abschnitte gegeneinander abgrenzen, sondern auch ihre zeitlich und räumlich koordinierte Aktivierung oder Deaktivierung kontrollieren. Diese Sequenzen sind die sogenannten SMARs - sie offerieren Bindungsstellen für Proteine und Proteinkomplexe, die in proprietärer oder modifizierter Form in die Regulation der Chromatin-Kondensation und Dekondensation, in die Bildung von Transkriptionsschleifen und die Formatierung von Chromatin-Domänen und damit in die Aktivierung von Promotoren und regulatorischen Proteinen eingreifen.
Wie seit geraumer Zeit bekannt ist, ist das Chromatin im Zellkern in unabhängigen, abgrenzbaren zellulären Bereichen konzentriert, die zugleich Struktur- und Funktionseinheiten des Genoms repräsentieren und heute allgemein mit Chromosomen-Kompartimenten assoziiert werden. In neueren Untersuchungen haben sich deutliche Hinweise darauf ergeben, dass die Regulation der Genexpression nicht allein von der Sequenz des Gens per se abhängig ist oder von den Promotoren, durch die es aktiviert werden kann, sondern auch von der räumlichen Anordnung des Gens (des Chromosoms, auf dem es sich befindet) im Zellkern und der Zugehörigkeit der regulatorischen Sequenzen und Elemente zu bestimmten Regionen des Genoms und des Kerns. Eine wichtige Rolle bei der Organisation der sagen wir tertiären107 - Struktur der DNA sollen die Kernmatrix und/oder das sogenannte Scaffold spielen, beides komplexe Gerüststrukturen108, die überwiegend aus Proteinen bestehen. In ihnen gibt es Bereiche - die wir weiter oben bereits als MARs oder SARs (SMARs109) kennengelernt haben - denen verschiedene Funktionen sowohl bei der DNA-Replikation als auch bei der Transkription zugeschrieben werden.
SMAR- Elemente wurden schon früh als DNA-Elemente registriert, die unterschiedliche Funktionen ausüben: sie wurden unter anderem mit der Transkriptionsaktivierung (Klehr et al., 1991; Bode et al. 1995) und der Langzeit-Expression von Genen in Verbindung gebracht (Dang et al., 2000).
Einen ausführlichen Einblick in dieses Thema gibt Frank O. Fackelmayer in seinem in Biospektrum (2000) 6, 441-444 veröffentlichten Aufsatz: Die Architektur des Zellkerns. Das folgende Zitat stammt aus diesem Aufsatz:
Auf Grund biochemischer und elektronenmikroskopischer Untersuchungen wird der Kernmatrix von vielen Autoren eine wichtige Rolle in der Zellkernarchitektur zugeschrieben. Die heutige Modellvorstellung geht davon aus, daß das Chromatin in Form von Schleifen organisiert ist, die im Durchschnitt zwischen 20000 und 80000 Basenpaare lang sind. Dadurch wird das Genom strukturiert, das heißt in überschaubar große Bereiche eingeteilt. Die Schleifen sind an ihrer Basis über bestimmte DNA-Sequenzen mit der Kernmatrix verbunden und dadurch so voneinander getrennt, daß jede Schleife eine unabhängige funktionelle Einheit oder Domäne darstellt. Im normalen Interphase-Zellkern sind Chromatinschleifen nicht direkt beobachtbar, wohl aber bei Lampenbürsten-Chromosomen oder polytänen Chromosomen bestimmter Amphibien- oder Insektenzellen. Bei diesen Sonderfällen werden Bereiche des Chromatins aufgelockert und als Schleife sichtbar, wenn die darauf liegenden Gene aktiviert werden und kondensieren wieder, wenn die Gene abgeschaltet werden. Auch bei höheren Zellen geht eine Aktivierung von Genen mit einer Auflockerung der Chromatinstruktur einher. Die aufgelockerte Region ist dabei meist größer als der kodierende Teil des Gens und entspricht der Chromatinschleife, auf der das Gen liegt.
[..]
Die DNA-Bereiche, die als Anheftungsstellen des Chromatins an die Kernmatrix dienen, werden je nach der Methode ihrer Isolierung als SARs (engl. scaffold attachment regions) oder MARs (engl. matrix associated regions) bezeichnet. SAR-Elemente liegen fast immer außerhalb proteinkodierender Bereiche. Sie haben eine Länge von 200-3000bp und zeichnen sich durch A+T-Reichtum aus, ohne daß jedoch eine einfache SAR-Konsensus-Sequenz bestimmt werden könnte. Dennoch werden SAR-Elemente artüberschreitend erkannt, was auf deren lebenswichtige Funktion für alle eukaryotischen Zellen hindeutet. Welche Funktion dies sein könnte, wird von verschiedenen Autoren unterschiedlich beurteilt. So zeigen SARs einen stimulierenden Einfluß auf die Aktivität benachbarter Gene, werden aber auch mit der Replikation des Genoms, DNA-Reparatur und Rekombination in Verbindung gebracht. Möglicherweise sind viele der gezeigten Funktionen von SARs auf deren architektonische Rolle zurückzuführen, weil die Funktionen nur im Kontext einer intakten Kernarchitektur ablaufen können.(Zitat Ende)
In den letzten Jahren sind eine Reihe von Proteinen entdeckt und untersucht worden, denen eine Funktion bei der Strukturierung oder Kompartimentierung von Chromatin und eine Beziehung zu SMARs zugeschrieben wird. Zu diesen Proteinen gehören auch die von der Gruppe Fackelmayer entdeckten Proteine SAF-A und SAF-B, sowie die Proteine SATB1 und SATB2, die im Labor Dr. Kohwi-Shigematsu entdeckt und charakterisiert wurden.
Beide Gruppen beschäftigen sich vor allem mit der Untersuchung von DNA-bindenden Proteinen, die für die funktionelle Architektur des Zellkerns von Bedeutung sind. Die von der Gruppe Fackelmayer entdeckten und charakterisierten SAF-A und SAF-B-Proteine (SAF= Scaffold Attachment Factors) binden spezifisch an bestimmte SMARs und haben nach Meinung der Gruppe Einfluß auf wichtige genetische Prozesse wie DNA-Replikation und Genexpression. Dafür sei eine Wechselwirkung mit anderen Proteinen des Zellkerns, mit DNA und vermutlich auch mit RNA notwendig. Neueste Ergebnisse aus dem Labor Fackelmayer lassen erkennen, daß diese Wechselwirkung zumindest bei SAF-A von einer speziellen posttranslationalen Modifikation des Proteins reguliert wird, der asymmetrischen Methylierung von Arginin-Seitenketten. Diese Modifikation ist auch von zahlreichen anderen nukleinsäure-bindenden Proteinen des Zellkerns bekannt, ihre funktionelle und strukturelle Rolle ist bis heute jedoch ungeklärt.
Das von der Gruppe um Dr. Kohwi-Shigematsu entdeckte Protein SATB1 scheint ein Molekül zu sein, das nur in bestimmten Zellinien vorkommt, so in Thymozyten, aktivierten T-Zellen und in einigen Vorläufern der genannten Zellen. Wenn die Expression dieses Proteins im Maus-Modell verhindert wird, wird die Regulation vieler Gene in Thymozyten und damit die Entwicklung von T-Zellen gestört. SATB1 soll weiter eine wichtige Rolle bei der Organisation der DNA in loops spielen, also in chromosomale Schleifen, die in unregelmäßigen Abständen an die Kernmatrix angeheftet sind: SATB1 soll dabei eine spezifische DNA-Sequenz erkennen, genannt BUR (Base Unpairing Region) die als charakteristisch für DNA-Bereiche gilt, in denen die DNA mit der Kernmatrix in Kontakt steht, also für die zuvor charakterisierten SMARs. SATB1 soll aktiv an der Bildung von Chromatin-Schleifen oder schlaufen (loops) beteiligt sein, indem es BUR-DNA-Sequenzen wie eine biologische Klammer miteinander verbindet. Darüberhinaus soll SATB1 auch mit der Rekrutierung von remodeling-Faktoren oder -komplexen wie dem NURD- und dem CHRAC/ACF-Komplex korreliert sein, was eine Erklärung für die Beobachtung bieten soll, dass SATB1 offensichtlich Einfluss auf die Expression von Genen hat.
Wenn man die zahlreichen zu SATB1 und neuerdings auch SATB2 vorliegenden Studien kritisch würdigt, muss man zu dem Schluss kommen, dass Proteine der SATB Familie mit hoher Wahrscheinlichkeit an DNA-Regionen (SMARs) binden, die für die Unterteilung eines Chromosomenbandes in Chromatin-Domänen charakteristisch sind und auch an der Bildung übergeordneter Chromatin-Strukturen an der Bildung von loops aus Chromatin-Subdomänen und an der Bildung von Chromomeren aus Chromatin-Domänen beteiligt sind. Chromomere die kondensierten Chromatin-Domänen also - stellen sich im SEM110 als 250-300nm-Strukturen dar eine definierte Anzahl von ihnen bilden nach meinen Computer-Analysen zunächst eine Transkriptionsgruppe und dann ein Chromosomenband. Chromatin-Domänen lassen sich, wie auf der Abbildung gezeigt, in Subdomänen einteilen, die wiederum von SMARs separiert werden. Und in Subdomänen schliesslich das werde ich in Teil III dieser Arbeit ausführlich besprechen habe ich eine mehr oder weniger grosse Zahl von spezifischen SMARs identifiziert, die REMA-Gene (und ALU-Gene) in Transkriptionseinheiten einteilen.
Verschiedene Klassen von SMARs strukturieren das Genom.
Sie sind mit Aktivierung und Repression von zwei Gen-Systemen verknüpft.
In der Literatur wird die Aktivierung und Repression von Genen allgemein mit der Wirkung von Isolator- und Enhancer-Elementen oder mit der Wirkung sogenanntre LCRs in Verbindung gebracht. Die genannten Elemente sind nur wenig spezifiziert: weder kennt man eine bestimmte Sequenz, noch besondere topologische Bedingungen, mit denen die Existenz dieser Regionen verknüpft werden könnte. LCRs sind zwar durch hypersensitive DNA-Regionen (HSS) gekennzeichnet - sie sind aber wie Enhancer und Isolatoren weder durch ihre exakt definierte Lage im Genom, noch durch eine Consensus-Sequenz, sondern vielmehr durch die Vielzahl der ihnen zugeschriebenen unterschiedlichen Eigenschaften und deren Auswirkungen auf die Genexpression charakterisiert.
In den folgenden Kapiteln will ich versuchen zu zeigen, dass Enhancer, Isolatoren und LCRs nichts anderes sind als spezifische SMARs, die das Genom in definierbare, funktionelle Bereiche unterteilen, die auf verschiedenste Art und Weise entweder direkt oder indirekt für die differentielle Genexpression verantwortlich zeichnen. Ich will versuchen, diese DNA-Regionen zwar nicht vollständig, aber doch so eingehend zu beschreiben, dass die lineare Gliederung und die logische Struktur der DNA erkennbar werden. Dabei wird deutlich werden, dass Enhancer-Regionen, LCRs und Isolator-Elemente verschiedene Klassen von SMARs darstellen, deren Funktionalität von der Bindung der zuvor erwähnten Proteine an definierte Sequenzen innerhalb dieser SMARs bestimmt wird. Die Bindung und Modifizierung dieser Proteine während bestimmter Phasen des Zellzyklus entscheidet darüber, ob die betreffenden SMARs einen reprimierenden, regulierenden oder aktivierenden Einfluss auf den jeweiligen DNA-Abschnitt haben.
Dabei konkurrieren verschiedene Protein-Gruppen in wechselnden Zusammensetzungen abhängig vom Typ der SMAR und vom genomischen Kontext um die gleichen Bindungsstellen111 und regulieren dadurch die Funktionalität der jeweiligen Chromatin-Region.
Meine Berechnungen und Analysen mittels IMPACD® lassen mich annehmen, dass es - wie oben bereits erwähnt - zumindest drei112verschiedene Arten oder Klassen von SMARs gibt, die mit jeweils unterschiedlich funktionellen DNA-Abschnitten verknüpft sind.
SMARs der Klasse I
werden durch Promotoren und Terminatoren (poly-A-Signalen) von metabolen Genen sowie von sense und antisensePromotoren von REMA- und ALU-Genen repräsentiert. Sie binden nur während der Transkription, also temporär an die Matrix oder an SMARs, die mit der Kernmatrix assoziiert sind. SMARS der Klasse I umfassen zwischen 5 und 30 Basenpaare.
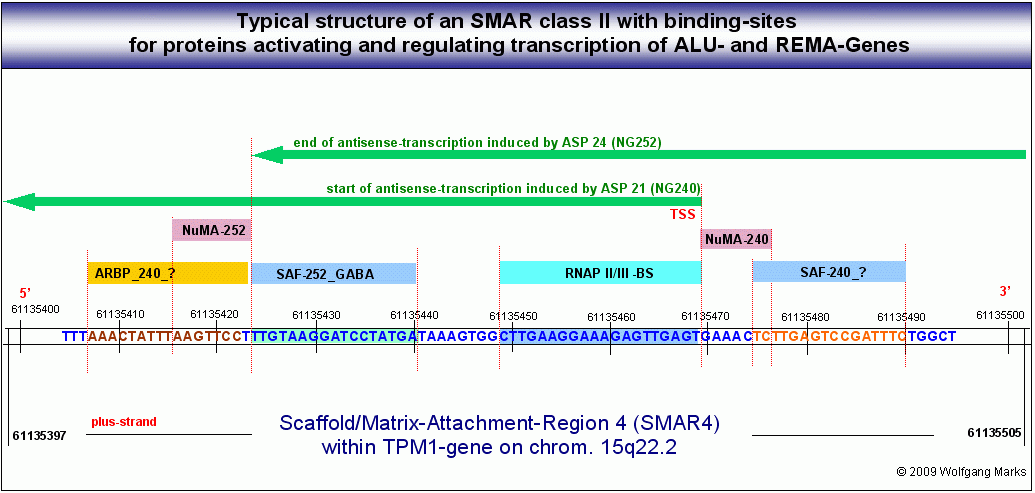
SMARs der Klasse II
zählen zwischen ca. 60 und ca. 200 Basenpaare. Sie unterteilen eine Subdomäne in Transkriptionseinheiten, die mit der Expression von REMA-Genen und ALU-Genen korreliert sind. Die Grafik XX zeigt eine mittels IMPACD® identifizierte SMAR II aus der Subdomäne 3.3 (siehe Abbildung). Diese SMAR II begrenzt ein ALU-Gen, das innerhalb jenes mehrfach erwähnten TPM1-Gens liegt, das ich später noch genauer untersuchen werde. Die Abbildung demonstriert die typische Struktur eines solchen Elements mit Bindestellen für verschiedene Proteine, die aktivierend, reprimierend oder regulierend in die Expression der konnektierten Gene eingreifen. Gene, die direkt von SMARs dieses Typs II reguliert werden, sind z. B. ALU-Gene oder REMA-Gene. Indirekt steuern diese SMARs des Typs II aber über die Transkriptionsfaktoren, die von ihren ALU-Genen kodiert werden, die Expression metabolischer Gene wie zum Beispiel die des TPM1-Gens. Darauf komme ich in Teil III unter anderem im Kapitel über die ALU-Gene noch zurück.
|
|
|
|
|
SMARs der Klasse III
sind nicht nur grössten SMARs im humanen Genom sie umfassen zwischen ca. 1500 und 4500 Basenpaare sondern auch die komplexesten, da die Eigenschaften dieses Typs von SMARs sich zellzykusabhängig verändern. SMARs der Klasse III begrenzen sowohl Chromatin-Domänen als auch Chromatin-Subdomänen innerhalb dieser Domänen. Ob sie als Domänen-SMAR oder als Subdomänen-SMAR in die zellulären Prozesse eingreifen, hängt davon ab, in welchem Stadium des Zellzyklus ihre Dienste gefragt sind oder in Anspruch genommen werden.
Allerdings unterscheiden sich Klasse III SMARs am Beginn einer Domäne in einem Punkt von den SMARs am Beginn einer Subdomäne: SMARs am Beginn einer Domäne, die ich REMAKEs genannt habe, enthalten eine Vielzahl von REMA-Boxen, die dockingstations für die zuvor beschriebenen remodeling-Maschinen darstellen und zugleich jeweils eine BS für einen origin of replication recognition complex (ORC) und damit zugleich den Origin Of Replication (ORI) repräsentieren. Ausserdem offerieren sie nach dem Ergebnis meiner Computer-Analysen Bindestellen für Proteine der SATB-, der SAF-, der HMGA-, HMGB-, der CTCF113- und der ARBP/MeCP2-, der HMGN- und der CTCF-Familie, die entweder in verschiedenen Kombinationen um die vorhandenen Bindestellen konkurrieren oder kumulativ binden. Manche dieser Proteine können wahrscheinlich nur dann an diese SMARs binden, wenn sie spezifisch modifiziert sind und/oder aktivierte Hormon-Rezeptorprotein-Komplexe gebunden haben. Proteine der ARBP/MeCP2-Gruppe zum Beispiel binden unter anderem an spezifisch modifizierte DNA-Nukleotide in den REMA-Boxen: dies kann die Methylierung von CpG-Dinukleotiden (MeCP2) sein, bei anderen Proteinen dieser Gruppe aber auch eine andere Art der Modifizierung114. Auch Helikasen, Ligasen, Polymerasen und Topo-Isomerasen binden in diesen Regionen entweder an Proteine, die die Bindung an eine REMA-Box vermitteln oder direkt an spezifische Sequenzmotive.
Ein grosser Teil dieser zuvor erwähnten Proteine bindet wahrscheinlich aber auch spezifisch an SMARs III, die eine Chromatin-Domäne in Subdomänen einteilen. An diese SMARs zwischen und vor Subdomänen, von denen ich einige identifiziert habe und später noch näher charakterisieren werde, binden vielleicht von ein oder zwei Ausnahmen abgesehen die gleichen Proteine wie an SMARS, die REMAKEs darstellen was nicht weiter verwundern sollte, da wie auf der Grafik gut zu erkennen ist - jeder REMAKE am Beginn einer Domäne auch zugleich den Beginn einer Subdomäne markiert.
Auch SMARs/REMAKEs am Beginn einer Domäne fungieren also während der Interphase des Zellzyklus als Subdomänen-SMARs. Sie sind in dieser Phase direkt mit der Expression metaboler Gene verknüpft. Sie enthalten, wie oben bereits gesagt, zum Teil die gleichen, zum Teil andere und zusätzliche Bindestellen wie SMARs der Klasse II, so zum Beispiel für HMGA-, HMGB-Proteine, CTCF-, SATB-, SAF-, ARBP-Proteine und spezifische Helikasen und Isomerasen.
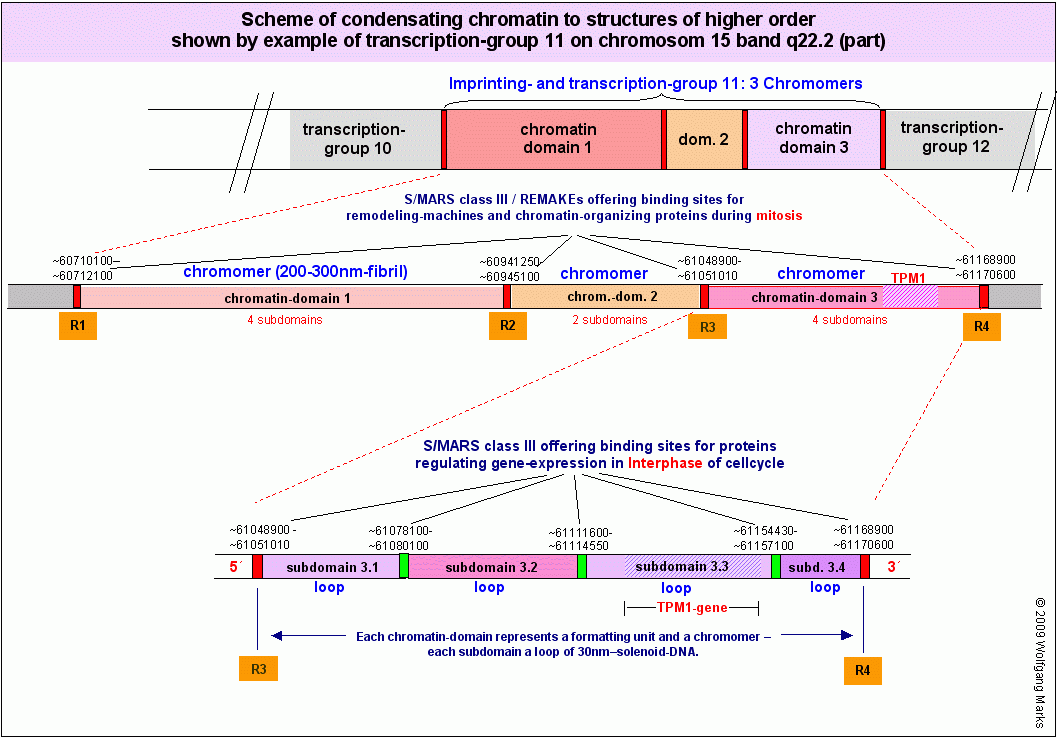
Obwohl die SMARS der Klasse III vor Domänen und Subdomänen also ähnliche Bindungsstellen offerieren, haben sie doch sehr unterschiedliche Funktionen: so haben zum Beispiel die SMARs R1 bis R4, die Chromatin-Domänen separieren wie wir auf den Grafiken weiter unten sehen - zum einen eine spezifische Funktion bei der (Neu)Formatierung115 dieser Domänen mit einer bestimmten Nukleosomengrösse (ein Prozess, der in zwei durch eine mitotische Teilung getrennten Zellzyklen abläuft), zum anderen eine ebenso spezifische Aufgabe bei der Steuerung der Expression der Gene der anliegenden Subdomänen in der G0- oder der Interphase des Zellzyklus. Die SMARs R1 und R4 haben darüberhinaus Kontrollfunktionen, die mit der Tatsache einhergehen, dass sie nicht nur Domänen- und Subdomänengrenzen markieren, sondern gleichzeitig auch die Grenzregion zwischen Transkriptionsgruppe 10 und 11 beziehungsweise 11 und 12 darstellen und damit über die genannten Funktionen hinaus auch noch Aufgaben bei der Kondensation respektive Dekondensation des Chromatins und der Regulation des Imprintings haben. Die angesprochenen SMARs der Klasse III haben also abhängig von der Phase des Zellzyklus und dem dadurch bestimmten Angebot an Bindungspartnern jeweils andere, sehr spezifische Funktionen, die dynamisch wechseln können.
Subdomänen-SMARs der Klasse III sind vermutlich identisch mit
Locus Control Regions (LCRs).
Locus Control Regions (LCR) die bereits vor längerer Zeit als wesentliche Steuerelemente der Genexpression identifiziert wurden - sind charakterisiert durch DNAse-hypersensitive Stellen und können weit (bis zu 100 kb und mehr) stromauf eines Gens liegen. Die Beziehungen zwischen LCRs und SMARs sind in vielen Punkten ungeklärt, da die bisher veröffentlichten Studien kein einheitliches Bild ergeben:
(Zitat)116 The role of MARs in the topological organization of the genome has suggested that they might have a role in transcriptional regulation. An emerging concept is that MARs organize the genome into discrete, autonomous regulatory domains. Studies in a number of systems support this model. The Drosophila melanogaster ftz, Sgs-4 (27), chicken lysozyme (28), and human ß-globin genes (29) are each flanked by MARs required for the insulation of transgenes from position effects (22,30). The prevalence of topoisomerase II sites at known MARs (4,31) and evidence linking active chromatin and torsional stress in higher eukaryotes (3234) suggests a mechanistic basis for such MAR-mediated functions. A number of MARs are also found in the proximity of transcriptional enhancers and may contribute to their ability to act over long distances. This enhancer facilitation may reflect sequestration of chromatin domains into specific subnuclear microenvironments and/or the regional modulation of chromatin accessibility. The most detailed evidence for the interaction of MARs with LCR elements and enhancers comes from studies of the intragenic mouse Igµ heavy chain LCR. The major enhancer element in this LCR is flanked by MARs (4,23). While these MARs have no appreciable effect on enhancer action in cell transfection studies (9,3537), they have been found to markedly augment Igµ enhancer action in transgenic mouse B-cells. They extend the distance over which the Igµ enhancer can influence chromatin accessibility, core histone acetylation, DNA demethylation, and transcription (9,11,12).
In contrast to the multiple studies documenting activity of MARs in gene expression, a number of studies indicate that these effects may not be universal (e.g. 38, reviewed in 39). For example, site-directed mutations of MARs flanking the murine ß-globin gene (40), the human A -globin gene (41,42) and the endogenous murine Igµ LCR (43) do not appear to alter expression of the associated promoters. Thus, the role of MARs may be specific to certain genes or particularly evident in specific experimental contexts.
The lack of MARs within the hGH LCR suggests that HSI,II activates the hGH-N transgene over an intervening distance of 15 kb by a MAR-independent mechanism. It has been established that this region of the LCR is a target for histone acetylation in a somatotrope nucleus (25), possibly through recruitment of histone acetyltrasferase complexes by the POU-homeodomain factor Pit-1 (21,44). This recruitment triggers the modification of a 32 kb domain of chromatin acetylation extending 5' to the most distal LCR determinant (HSV; 32 kb) and 3' to the hGH-N promoter. This modification, targeted to HSI,II and subsequently spread throughout an extensive domain, may be sufficient to mediate hGH-N gene activation. Thus, the hGH LCR may utilize a mechanism of long-range gene activation that obviates the need for MAR function at this locus. (Zitat Ende)
Die Schwierigkeiten bei der Unterscheidung von SMARS und LCRs ergeben sich im Wesentlichen daraus, dass SMARs der Klasse III wie zuvor ausführlich erläutert -unterschiedliche Funktionen in Abhängigkeit vom Zellzyklus ausüben. Während der Mitose fungieren sie als Andockstationen für Remodeling-Maschinen, in der Interphase als Locus Control Regions und damit als Bindestellen für verschiedene Proteine und Faktoren, welche die Genexpression aktivieren und modulieren. Ob eine SMAR sich als LCR oder als REMAKE geriert, ist also vom Zeitpunkt der Betrachtung oder der Untersuchung abhängig.
Es scheint aber schon jetzt evident, dass sowohl die Funktionen, die mit den LCRs verknüpft sind als auch ihre bis jetzt beschriebenen Charakteristika mit denen der SMARs der Klasse III im zuvor beschriebenen zellulären Kontext vollständig kongruent sind. Es kann deshalb für mich keinen vernünftigen Zweifel daran geben, dass die von mir entdeckten und erstmals charakterisierten SMARs der Klasse III identisch mit den apostrophierten locus control regions sind.
Zitat: Locus control regions (LCRs) are operationally defined by their ability to enhance the expression of linked genes to physiological levels in a tissue-specific and copy number-dependent manner at ectopic chromatin sites. Although their composition and locations relative to their cognate genes are different, LCRs have been described in a broad spectrum of mammalian gene systems, suggesting that they play an important role in the control of eukaryotic gene expression. The discovery of the LCR in the beta-globin locus and the characterization of LCRs in other loci reinforces the concept that developmental and cell lineage-specific regulation of gene expression relies not on gene-proximal elements such as promoters, enhancers, and silencers exclusively, but also on long-range interactions of various cis regulatory elements and dynamic chromatin alterations.117
Zusammenfassung
SMARs können Chromatin-Domänen separieren oder Subdomänen voneinander trennen - sie unterteilen Subdomänen in Transkriptionseinheiten für REMA-Gene und ALU-Gene118und weisen erhebliche Größenunterschiede auf. Letztlich aber werden die verschiedenen Klassen von SMARs durch die Proteine oder Proteinkomplexe (remodeling-Maschinen) charakterisiert, die in diesen Regionen zellzyklus-abhängig an die DNA (und die Matrix) binden und dadurch die Funktionalität der von den SMARs umschlossenen oder gegliederten Regionen oder die Funktionalität der SMARs selbst bestimmen.
Der folgende Versuch einer Klassifizierung, deren Systematik im Verlauf der folgenden Ausführungen hoffentlich deutlich werden wird, beruht auf den von mir bis heute identifizierten humanen SMAR-Sequenzen. Nur SMARs vom Typ III binden konstitutiv an die Matrix, alle anderen SMARs nur während der Transkription, also temporär. Dies wird unter anderem auch Thema in Teil III dieser Arbeit sein.
|
SMAR
class I
|
|
SP/ASP z.B. von ALU- und REMA-Genen, Promotoren u.Terminatoren von Genen binden temporär an die Matrix oder an SMARs.
|
|
~5-
30 bp
|
|
SMAR
class II
|
In REMA- u. ALU-Genen, definieren Transkriptionseinheiten dieser Gene Grösse hängt von Zahl der NuMA-BS ab. Binden nur temporär an die Matrix.
|
|
|
SMAR
class III
|
Begrenzen Chromatin-Domänen (REMAKEs) und Chromatin-Subdomänen. Funktion ist zellzyklusabhängig. REMAKEs sind konstitutiv an die Matrix gebunden.
|
~1500-
4500 bp
|
|
|
|
|
|
Transkriptionsgruppen Domänen Subdomänen
und ihre Beziehung zu Chromosomenmodellen.
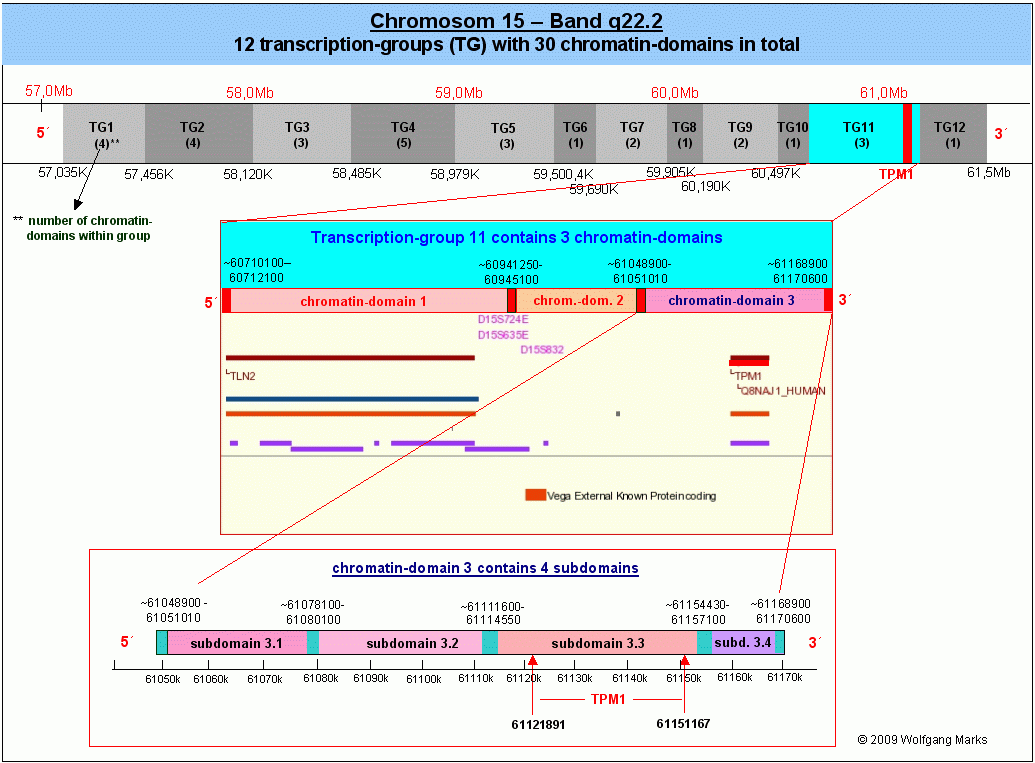
Die Abbildung unten links zeigt das Band q22.2 des Chromosoms 15. NCBI schreibt diesem Band dreissig bzw. 40 (Struktur-) Gene zu, was den tatsächlichen Verhältnissen ziemlich nahe kommen dürfte (diese Zahl hängt letztlich davon ab, welche Sequenzen man zu den Genen119 zählt (REMA-Gene, UsnRNA-Gene, ALU-Gene, andere interspergierte repetitive Elemente?). Im Band 15q22.2 habe ich mit Hilfe von IMPACD® zwölf Transkriptionsgruppen definiert, die ich auf der Abbildung mit TG1 bis TG12 bezeichnet habe. Diese 12 Transkriptionsgruppen repräsentieren insgesamt 30 Chromatin-Domänen.120 Die Transkriptionsgruppe 11, in der das TPM1-Gen liegt, habe ich genauer untersucht und durch SMARs in Chromatin-Domänen und darüberhinaus in Subdomänen eingeteilt. Die Subdomäne 3.3 in der sich das TPM1-Gen befindet - werde ich in Teil III dieser Arbeit im Detail analysieren.
 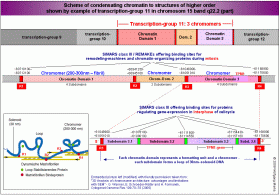
Wenn über Struktur und Organisation der DNA gesprochen wird und über ihre Einteilung in funktionelle Bereiche, sollte stets im Auge behalten werden, dass diese funktionellen Bereiche sehr unterschiedliche Aufgaben zu erfüllen haben. Zum einen müssen sie gewährleisten, dass ein etwa einen Meter langes Molekül durch Kondensation, Schraubung und Faltung zu einem nur wenige Mikrometer langen Chromosom gepackt werden kann, zum anderen müssen sie nach Dekondensation zum aktiven Chromatin (das auch inaktive Bereiche enthalten kann) die kontrollierte und koordinierte Expression von Genen gewährleisten, die über das ganze Chromosom (und gegebenenfalls andere Chromosomen) verstreut sein können.
Während bei der Kondensation des Chromatins zum Chromosom konstante Bindestellen in den REMA-Boxen der REMAKEs-SMARs der Klasse III für Proteine wie SATB1 und SAF-A/B, HMGA-, HMGB-, CTCF-, ARBP-Proteine und andere zur Bildung von Solenoid-Strukturen, Solenoid-Schlaufen/loops, Chromomeren und Chromosomenbändern genutzt werden, sind die Chromatin-Schlaufen aus der 10nm-Elementarfibrille also der unverdichteten Nukleosomenkette, die den transkriptionsaktiven Zustand darstellt - wie wir später noch sehen werden, nicht konstant mit definierten Matrix-Regionen verbunden, sondern abhängig von Zelle und Differenzierungsstadium (von der Nukleosomengrösse) dynamisch mit den REMAKEs/SMARs der Klasse III und definierten Promotoren sowie Terminatoren verbunden, die die Bildung eines Primärtranskripts und dessen Prozessierung determinieren.
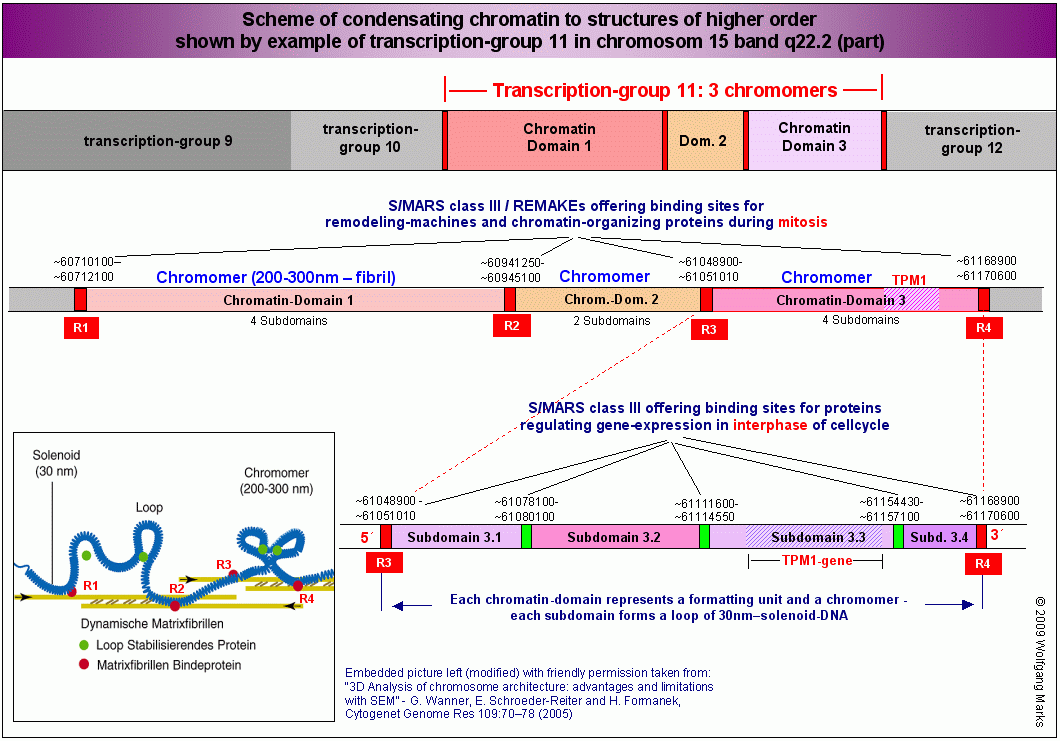
Wie auf der Abbildung Scheme of condensating chromatin to structures of higher order (oben rechts) und der Übersicht über das Band 15q22.2 zu sehen ist, sind weder Chromatin-Subdomänen, noch Chromatin-Domänen bzw. Chromomere Einheiten gleicher Größe: damit kommen zumindest für mich Chromosomenmodelle, die eine regelmäßige Periodizität von Chromatin-strukturen erfordern, nicht in Betracht. Das betrifft sowohl das sogenannte radiale Schlaufenmodell (Adolph und Kreisman, 1985), als auch das Mehrfachschrauben-modell (multiple coiling model) von Taniguchi und Takayama (1968) und die sogenannten helikalen Chromosomenmodelle (scaffold model) von Filipsiki (1990), Laemmli (1994) und Stack und Anderson(2001).
|
|
|
|
|
|
|
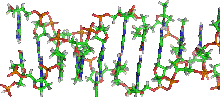
Das einzige mir bekannte Modell, das mit meinen Beobachtungen und Berechnungen zur Struktur und Organisation der DNA vollständig übereinstimmt, ist das neue Matrix-Fibrillen-Modell von Wanner und Formanek. Die Abbildungen wurden mit freundlicher Genehmigung entnommen aus: 3D Analysis of chromosome architecture: advantages and limitations with SEM. G. Wanner, E. Schroeder-Reiter and H. Formanek, Department of Biology I, Ludwig-Maximillians-Universität München, Munich (Germany) Cytogenetic and Genome Research 109:7078 (2005). Bild WA1 wurde von mir verändert - ich habe REMAKEs und Subdomänen-SMARs gekennzeichnet.
|
|
Das einzige mir bekannte Modell, das mit meinen Beobachtungen und Berechnungen vollständig übereinstimmt, ist das bereits erwähnte und in den Abbildungen dokumentierte neue Matrix-Fibrillen-Modell von Wanner und Formanek, das eine ebenso einfache wie einleuchtende - durch umfangreiche SEM-Analysen gestützte - Erklärung bietet für den Weg, den die Zelle beschreitet, wenn sie ihre DNA zur Transportform komprimiert. Wenn man sich die Zeichnung Scheme of condensating chromatin to structures of higher order genauer ansieht, werden die Übereinstimmungen hoffentlich deutlich.
Die Gliederung der DNA in Ordnungsstrukturen (die genomische Syntax) zeigt sich also auch oder gerade in der Transportform der DNA, dem Chromosom. Die je nach Präparations- und Färbemethode unterschiedliche, aber strukturell einheitliche Anfärbung bestimmter Bereiche des Metaphase-Chromosoms ist nach meiner festen Überzeugung korreliert mit ebenso einheitlichen chemischen Modifikationen dieser Abschnitte. Denn das System von genomischen Schlüsseln, das ich in einem der vorhergehenden Kapitel skizziert habe, muss sich während der Phylogenese nach inhärenten logischen Prinzipien entwickelt haben, da seine Effekte auf die DNA wie ich später zeigen werde mathematisch beschreibbar sind. Demnach muss es auch im System der Histon- und DNA-Modifikationen eine logische Ordnung geben, die sich in jeder Struktur, also auch in Chromosomenbändern während der Pro- oder Metaphase unter geeigneten Bedingungen chemisch und/oder optisch determinieren lässt.
Lassen Sie mich an dieser Stelle kurz erinnern, was wir über das chemische Verhalten von Histonen wissen:
Die Histonklassen H2A/H2B auf der einen, H3 und H4 auf der anderen Seite können unab-hängig voneinander Aggregate bilden: bei Assoziationsversuchen aggregieren die arginin-reichen Histone H3 und H4 zum Tetramer H3(2x) - H4(2x), die schwach lysinreichen Histone H2A und H2B bilden zunächst ein Dimer und anschließend ebenfalls das Tetramer H2A(2x) - H2B(2x). Dies weist unmissverständlich daraufhin, daß das Oktamer eine a priori zweigeteilte oder viergeteilte Struktur besitzt, deren Ursache nicht in chemischen Modifikationen von spezifischen Aminosäuren der vier beteiligten Histonklassen zu suchen, sondern systemimmanent und auch ein Hinweis darauf ist, dass die Organisation der DNA in nukleosomale Strukturen nicht mit einem Histon-Tetramer oder -Oktamer, sondern wie weiter oben schon erwähnt - mit einem H3-Dimer begann.
Wie ich noch zeigen werde, findet Transkriptions-Initiation und Regulation der sogenannten Struktur- oder metabolen121 Gene auf der Ebene von Chromatin-Domänen und -Subdomänen statt bei der Initiation der Transkription spielt die Dissoziation von Histonen, wie sie weiter unten beschrieben wird, die entscheidende Rolle. Spezifische Modikationen an definierten Histonen entscheiden letztlich darüber, wie die Histone und welche Histone sich von der DNA lösen und damit auch darüber, welcher Strang transkribiert wird.
Für die Bildung der Chromosomen-Struktur, für die Bildung der Muster, die sich in der Metaphase als Bänderung der Chromosomen sichtbar machen lassen, sind also koordinierte, einem gemeinsamen System folgende Modifikationen an definierten Histonen eben das Chromatin-remodeling - verantwortlich. In den folgenden Kapiteln werde ich versuchen, dieses System näher zu beschreiben und neu zu strukturieren.
|
|
104Dieses System kann nicht das Hox-System sein. Wie ich später zeigen werde, regulieren diese Proteine die koordinierte Expression von Genen auf einer anderen Ebene.
105Oder anders ausgedrückt: eine remodeling-machine für die NG 236-S, die aus remodeling-Faktoren im Chromosomenband 15q22.31 generiert wurde, könnte sich (muss sich aber nicht) von der im Chromosomenband 15q22.2 gebildeten für die gleiche NG 236-S in der Zusammensetzung der Remodeling-Faktoren vielleicht nur geringfügig, aber eben doch unterscheiden.Mit grosser Wahrscheinlichkeit sind Chromosomenbänder und damit auch die dort kodierten remodeling-Maschinen mit Stammzellreihen korreliert.
106Zahlen jeweils ohne novel genes also erst kürzlich entdeckte und nicht klassifizierte Gene
107Primäre Struktur: die Abfolge der Basen und die daraus resultierende Helix-Form der DNA - Sekundäre Struktur: die Organisation in Nukleosomen einer definierten Größe (spacing) - Tertiäre Struktur: Organisation der formatierten DNA in Subdomänen, Domänen, Transkriptionsgruppen, Bänder und Chromosomen.
108Scaffold und Kernmatrix sind wahrscheinlich präparationsabhängig unterschiedliche Erscheinungsformen der gleichen zellulären Struktur.
109SAR- und MAR-Elemente werden heute allgemein unter dem Begriff SMAR zusammengefasst, weil sich gezeigt hat, dass es sich bei den bislang identifizierten MAR- und SAR-DNA-Regionen um Sequenzen mit gleichen oder sehr ähnlichen funktionellen Eigenschaften handelt.
110SEM: Scanning Electron Microscope = Raster-Elektronen-Mikroskop
111Siehe hierzu auch die SMAR zwischen Subdomäne 3.2 und 3.3. Details werden erst in Teil III erörtert.
112Letztlich hängt die Anzahl der SMAR-Klassen oder Typen davon ab, wie man diese Elemente definiert. Wenn man SMARs als DNA-Elemente beschreibt, die permanent oder temporär an die Kernmatrix (oder das Scaffold) gebunden sind und einen Einfluss auf die Genexpression, -regulation oder die Organisation der DNA in funktionelle Abschnitte haben, muss man in Konsequenz die SP/ASP-Promotor-Sequenzen von REMA- und ALU-Genen und die Promotoren anderer Gene ebenfalls zu den SMARs rechnen, weil auch diese bei der Bildung eines Transkriptionskomplexes zumindest temporär an die Kernmatrix binden.
113Abgeleitet von CCCTC binding factor man nahm ursprünglich an, dass das Protein präferenziell an CT-reiche Motive bindet. Diese Annahme erwies sich aber als falsch.
114Siehe auch Teil III: Das ARBP/MeCP2-Protein.
115Deswegen habe ich sie REMAKEs genannt. Dazu später mehr.
116The human growth hormone locus control region mediates long-distance transcriptional activation independent of nuclear matrix attachment regions; Nucl. Acids Res., Vol. 29, No. 16. (15 August 2001), pp. 3356-3361; by Brian M Shewchuk, Nancy E Cooke, Stephen A Liebhaber.
117Locus control regions; Li Q, Peterson KR, Fang X, Stamatoyannopoulos G.; Blood. 2002 Nov 1;100(9):3077-86.
118Siehe die Kapitel ALU-Gene 1 und ALU-Gene 2.
119Der Begriff des Gens muss wohl wieder einmal neu definiert werden.
120Im humanen Genom gibt es nach meiner Berechnung ca. 26.500 Chromatin-Domänen. Die durchschnittliche Größe einer Chromatin-Domäne liegt damit bei ca. 115.000 Basenpaaren.
121Gene, die am allgemeinen Metabolismus der Zelle beteiligt sind.
|
|
|