|
|
 |
|
|
|
Wolfgang Marks: Die Formatierte DNA
|
|
|
Der Weg zur zellspezifischen mRNA:
die Transkription.
Sind Architekturproteine Teil des Transkriptosoms?
Wenn man die Primärliteratur studiert, die sich mit dem Transkriptionsprozess im allgemeinen und spezifisch mit der Bildung des Transkriptionskomplexes, der Initiation der Transkription und ihrer Terminierung befasst, fällt auf, dass von den Architekturproteinen, die ich als wesentliche Bestandteile des Transkriptosoms genannt habe, mit Ausnahme eines HMGB-Proteins (HMGB1 identisch mit dem negativen Cofaktor NC1) nirgendwo die Rede ist.
Dies kann natürlich damit zusammenhängen, dass die meisten, wenn nicht alle Beobachtungen des Transkriptionsgeschehens entweder in vitro, an standardisierten und/oder immortalisierten Zellkulturen oder an der Bäckerhefe S. cerevisiae erfolgten, aber auch damit, dass den Untersuchenden der konkrete genomische Kontext, in dem diese Prozesse ablaufen und die in dieser Arbeit beschriebene systematische und logische Struktur der DNA und des Genoms nicht bekannt sein konnten.
Auch die Tatsache, dass bis heute nur einzelne oder einige wenige Mitglieder einer bestimmten Familie von Architekturproteinen entdeckt wurden, macht ihre Identifizierung in theoretischen oder artifiziellen Analysesystemen wahrscheinlich sehr schwierig oder ganz unmöglich.
Ein weiterer Grund könnte darin liegen, dass die von mir postulierten Akteure der genannten Proteingruppen ihre Konformation bei der Assemblierung der Transkriptionsmaschine ändern oder mit anderen Proteinen neue Komplexe mit neuen Funktionen bilden und dadurch nicht mehr identifiziert werden können.
Vielleicht auch lösen sich die Architekturproteine nach Bildung des Transkriptosoms wieder von der Matrix und der DNA und sind deshalb nicht mehr aufzufinden.
Mediatoren und Mediatorkomplexe: ein komplexes Problem und
ein Weg zu seiner Lösung.
Halten wir uns noch einmal den Weg vor Augen, der zur Bildung einer ganz bestimmten Transkriptionschleife, zum Beispiel der Schleife 13 führt. An den verschiedenen Schritten, die zur Installation dieser Schleife beitragen, sind Architekturproteine verschiedener Familien beteiligt, deren Synthese durch Hormone induziert wird, die zu einer Hormonkaskade gehören (Beispiele:TRH/TSH/y-Lipotropin - TRH/TSH/T4 - CRH/ACTH/Cortisol)243.
Die Zusammensetzung der sich daraus ergebenden, an die SMAR/Matrix gebundenen Komplexe aus verschiedenen Architekturproteinen kann demnach, abhängig vom Zelltyp und der Entwicklungsstufe, in der sich die Zelle befindet, sowohl was die Anzahl der Proteine als auch ihr Molekulargewicht betrifft, sehr verschieden sein die Komposition dieser Aggregate ist aber nicht willkürlich, sondern folgt bestimmten Regeln, die ich hoffe, in dieser Arbeit deutlich gemacht zu haben.
Auch die Zusammensetzung der bereits des öfteren erwähnten Mediator-Komplexe hat sich offensichtlich während der Evolution der Lebewesen nach bestimmten Regeln und Gesetzen entwickelt. Von den heute bekannten ca. 30 Untereinheiten von Mediator-Komplexen in Mammaliern sind 22 von der Hefe bis zum Menschen allerdings zumeist nur sehr schwach konserviert. Bestimmte Mediatoren, der MED25 zum Beispiel, kommen in menschlichen Zellen, aber nicht in der Hefe vor. Offensichtlich wurden das Spektrum an Mediatoren oder Mediator-Untereinheiten im Verlauf der Evolution erweitert.
Über Mediatoren ist in den vergangenen Jahren intensiv geforscht worden. Im Zuge dieser Arbeit ist eine Fülle von Untersuchungsergebnissen und Informationen zusammengetragen worden, die für sich genommen zwar noch kein erkennbares Bild, aber zusammen mit dem vor mir hier Vorgetragenen einen Sinn ergeben.
Da allein die Darstellung der verschiedenen bis heute identifizierten Mediatoren und Mediator-Komplexe und die ihrer Beziehungen untereinander ein ganzes Buch füllen würde, verweise ich an dieser Stelle auf eine Aufsatzsammlung244, die eine sehr gute Übersicht über den Stand der Forschung bietet insbesondere das Kapitel 4 General Cofactors: TFIID, Mediator and USA, das von Mary C. Thomas und Cheng-Ming Chiang geschrieben wurde, ist im aktuellen Zusammenhang von besonderem Interesse. Wer also tiefer in die Welt der Mediatoren und Mediator-Komplexe eindringen will, dem sei die Lektüre dieses Buches empfohlen.245
Wer meinen Ausführungen bis hierher gefolgt ist, wird es wissen oder ahnen:
Der überwiegende Teil der Mediatoren und Mediatorkomplexe muss aus Architekturproteinen bestehen.
Theoretisch könnte ich alle bis dato bekannten genetisch definierten Mediatoren mittels computational analysis via IMPACD® mit einem bestimmten Architekturprotein in Zusammenhang bringen - zumindest dann, wenn es sich bei dem betreffenden Mediator tatsächlich um einen solchen handelt. Der zeitliche Aufwand dazu wäre zwar überschaubar, aber im Moment von mir nicht zu leisten. Ich habe mich deshalb auf einige wenige, zufällig herausgegriffene Mediatoren beschränkt. Bei diesen wenigen allerdings habe ich bereits bemerkt, dass die Mediatoren offensichtlich alle aus den gleichen Komplexen oder aus Zellkulturen der gleichen Zell-Linien isoliert und extrahiert wurden. Anders ist nicht erklärbar, dass bei den in der Tabelle aufgeführten verschiedenen Architekturproteinen sowohl das Formatierungshormon MSH-RH als auch das primäre Transkriptionshormon ß-MSH immer wieder in Erscheinung treten.
Die einzelnen Komponenten von Mediatorkomplexen mittels IMPACD® zu identifizieren, wäre nur dann möglich, wenn genaue Informationen über die Zelle, aus der sie isoliert wurden und das Entwicklungsstadium dieser Zelle vorlägen. Da es sich bei diesen Zellen aber fast immer wie bereits angemerkt um Zellkulturen handelt, die von einigen wenigen Zell-Linien abstammen, stünde der Aufwand wahrscheinlich in keinem Verhältnis zum (mehr oder weniger vorhersehbaren) Ergebnis.
Ich habe deshalb ausser dem Mediator-Komplex PC2246 keinen Mediatorkomplex näher untersucht und den Komplex PC2 auch nur im Hinblick auf seine putative Zusammensetzung, ohne eine Zuordnung zu spezifischen Hormonen vorzunehmen.
Der PC2 besteht aus 15 verschiedenen Komponenten, von denen nach meinen Analysen 9 zu den Architekturproteinen gehören müssen. PC2 sollte sich demnach aus spezifischen Proteinen der HMGB-, HMGN-, SAF-, SATB- und ARBP-Familie zusammensetzen. Die anderen Bestandteile sollten eine Topo-Isomerase, zwei spezifische Hormon-Rezeptorproteine, vier Proteine der NuMA-Familie und 3 weitere Proteine sein, die ich nicht identifiziert habe.
Die Zuordnung einiger genetisch eindeutig definierter Mediatoren zu Architekturproteinen meines Systems zeigt die folgende Tabelle. Vielleicht werde ich diese Aufstellung zu einem späteren Zeitpunkt erweitern.
|
|
|
|
Mediatoren und ihre Beziehung zu Architekturproteinen
|
|
Mediator1
|
Protein Nr.
|
Identic with
|
FH
|
PTH
|
STH
|
|
MED4 (ARC36)
|
Q9NPJ6
|
ARBP244-PGB2
|
MSH-RH
|
ß-MSH
|
PGB2
|
|
MED6 (hMed6)
|
NP_005457
|
CTCF-244-MSH
|
MSH-RH
|
ß-MSH
|
-
|
|
MED15 (ARC105)
|
NP_001003891
|
NuMA-244
|
MSH-RH
|
-
|
-
|
|
MED20 (hTRFP)
|
NP_004266
|
Horm.-Receptor
|
MSH-RH
|
ß-MSH
|
PGB2
|
|
MED26/CRSP70
|
NP_004822
|
SATB-244-MSH
|
MSH-RH
|
ß-MSH
|
-
|
|
MED31 (hSOH1)
|
NP_057144
|
HMGN-244
|
MSH-RH
|
-
|
-
|
|
FH=Formattinghormone; PTH=Primary Transcript.-Hormone; STH=Second.Transcript.-Hormone
1Homo Sapiens
|
|
|
|
Die Transkription.
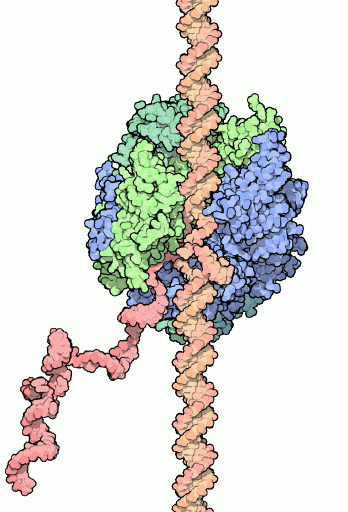
Die grundsätzliche Struktur des DNA RNAP II mRNA-Komplexes während des Ablesevorganges ist weitgehendst bekannt (Abbildung aus Wikipedia).
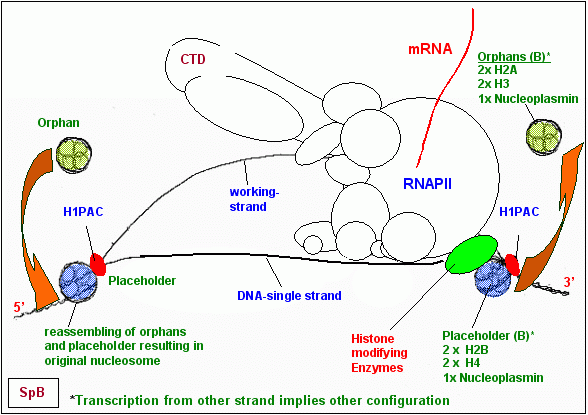
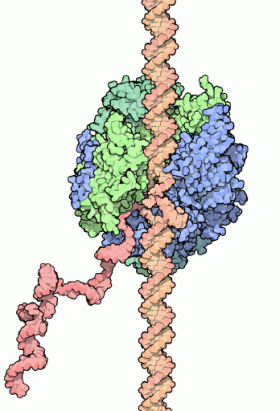 Bevor die RNAPII mit der Ablesung eines der beiden DNA-Stränge beginnen kann, hat die Zelle zunächst zwei Probleme zu lösen. Problem 1 besteht darin, dass ein mit Nukleosomen besetzter DNA-Strang nicht transkribiert werden kann. Problem 2 ist mit Problem 1 verknüpft und besteht aus dem Histon H1, das an der Schnittstelle zwischen DNA und Histon-Oktamer eine Art Klammer zwischen Oktamer und DNA bildet und die Öffnung des DNA-Doppelstrangs und damit auch die Deassemblierung der Histon-Oktamere zuverlässig verhindert. Die Lösung dieses Problems habe ich weiter oben bereits besprochen: sie besteht in der Verdrängung von H1 durch den H1PAC in einer bestimmten Phase der Transkriptionsschleifen-Bildung. Die Rolle, welche die H1PAC-Bestandteile Ubiquitin und Nukleoplasmin in diesem Komplex spielen, bedarf allerdings noch der näheren Erläuterung. Bevor die RNAPII mit der Ablesung eines der beiden DNA-Stränge beginnen kann, hat die Zelle zunächst zwei Probleme zu lösen. Problem 1 besteht darin, dass ein mit Nukleosomen besetzter DNA-Strang nicht transkribiert werden kann. Problem 2 ist mit Problem 1 verknüpft und besteht aus dem Histon H1, das an der Schnittstelle zwischen DNA und Histon-Oktamer eine Art Klammer zwischen Oktamer und DNA bildet und die Öffnung des DNA-Doppelstrangs und damit auch die Deassemblierung der Histon-Oktamere zuverlässig verhindert. Die Lösung dieses Problems habe ich weiter oben bereits besprochen: sie besteht in der Verdrängung von H1 durch den H1PAC in einer bestimmten Phase der Transkriptionsschleifen-Bildung. Die Rolle, welche die H1PAC-Bestandteile Ubiquitin und Nukleoplasmin in diesem Komplex spielen, bedarf allerdings noch der näheren Erläuterung.
Die prätranskriptionelle Ubiquitinierung von Histonen ist mit der Definition des aktiven Stranges und dem Nukleosomen-Splitting verknüpft.
Dass Ubiquitinasen auf dem Wege der Ubiquitinierung von Histonen in die Transkription eingreift, ist in verschiedenen Arbeiten nachgewiesen worden, ohne dass die Rolle dieser Ubiquitinierung vollständig aufgeklärt werden konnte. Robzyk, Recht et al.247 konnten belegen, dass in Sacch. cerevisiae das Protein Rad6 die Expression des Gens der Argininosuccinat-Synthase (ARG1) reguliert. In Abwesenheit von Rad6 wird ARG1 transkribiert ist das Protein im Zellkern vorhanden, wird die Transkription unterdrückt. Die Inaktivierung des ARG1-Gens soll auf der Ubiquitinierung von H2B beruhen, die durch Rad6 vermittelt wird. Sun and Allis248schliesslich gelang der Nachweis, dass es die Bindung eines Ubiquitin-Moleküls an das Lysin 123 des Histons H2B ist, die zur Methylierung von Lysin 4 in Histon H3 und damit zur Stillegung des Gens führt.
Dass die Ubiquitinierung von Histonen eine von vielen verschiedenen Modifikationen ist, die von Remodeling-Maschinen während der Mitose an den Histonen vorgenommen werden können, habe ich im Kapitel Chromatin-Remodeling bereits ausführlich diskutiert. Die Ubiquitinierung eines oder mehrerer Histone sind aber sicher nicht die einzigen Modifikationen (wenn auch vielleicht die entscheidenden), die zur Formatierung einer Chromatin-Domäne mit der NG 208 bzw. 150 und damit zur Stilllegung der Domäne beitragen.
Ich vermute deshalb, das die Bindung eines spezifischen H1PACs und die damit verbundene prätranskriptionelle Ubiquitinierung von definierten Histonen des Oktamers die Art der Deassemblierung der Oktamere determiniert, also mit der Definition des aktiven, des zu transkribierenden DNA-Stranges verknüpft ist.
Ich hatte 1990 postuliert, dass die Histon-Oktamere nach einem bestimmten Schema deassembliert werden, sich also nach einem bestimmten Schema teilen dabei sollte der zu transkribierende Strang gänzlich frei von Histonen sein am Gegenstrang dagegen definierte Komponenten der Histon-Oktamere verbleiben. Den Platz der vom Gegenstrang dissoziierten Histone sollten ein zellspezifischer HRPK und Nukleoplasmin einnehmen. Die dissoziierten Histone die Orphans sollten sich ebenfalls mit einem zellspezifischen HRPK und Nukleoplasmin verbinden und solange in diesem Zustand, in der Warteschleife bleiben, bis sie nach Austritt der DNA aus dem RNAPII-Komplex wieder im Austausch gegen ihre Platzhalter an ihre angestammte Position zurückkehren können.
Auch aus heutiger Sicht der Dinge erscheinen meine Hypothesen zum Ablauf der Strangtrennung und der Deassemblierung der Histone grundsätzlich folgerichtig und logisch. Lediglich bei den Histon-Klassen, die an der DNA verbleiben oder von ihr dissoziieren, habe ich aus aktuellem Anlass Änderungen vorgenommen. Details können der untenstehenden Tabelle und der Grafik entnommen werden.
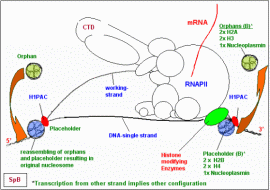
Meine Vorstellungen vom Prozess der Nukleosomen-Deassemblierung hatte ich bereits 1990 in einer Skizze festgehalten die Zeichnung links ist eine modifizierte Version des Originals von 1990.
Wie ich an anderer Stelle ausführlich diskutiert habe, enthalten Nukleosomen-Cores in Abhängigkeit vom epigenetischen Programm der Zelle sowohl canonische Histone als auch spezifische Histon-Varianten, zum Beispiel Schlusstein-Histone, die anzeigen, dass das Formatierungs-potential einer bestimmten Domäne erschöpft ist. In der Tabelle muss also gegebenenfalls ein canonisches Histon durch eine Histon-Variante ersetzt werden.
Welcher DNA-Strang transkribiert wird, ist aber letztlich entschieden worden, lange bevor sich die RNAPII nach Bildung des Transkriptosoms an die Arbeit macht: wie ich im Kapitel über das Chromatin-remodeling ausgeführt habe, sind für die Auswahl des zu transkribierenden Stranges in einer gegebenen Zelle nicht allein der Core-Promotor des zu transkribierenden Strukturgens, sondern auch die spezifischen Modifikationen verantwortlich, die ein definierter ATP-abhängiger Remodeling-Komplex an der DNA und den Histonen in einer vorhergehenden Phase der Zellentwicklung während einer vorhergehenden differentiellen Mitose - vorgenommen hat. Auch die Organisation des Genoms in Transkriptionsgruppen spielt bei der Festlegung des aktiven Stranges eine Rolle: daran sind zunächst (etwa vom 5. Tage an) HMGA-Proteine, ab dem 80. Tag in bestimmten Phasen der Ontogenese auch SATB- und CTCF-Proteine beteiligt.
Das Nukleosomen-Splitting.
Das Chromatin ist also im Augenblick der Transkription auf eine definierte Art und Weise organisiert und modifiziert. Auf dieser Plattform und nur auf dieser Plattform kann der zuvor beschriebene Prozess der gerichteten Nukleosomen-Deassemblierung ablaufen.
|
Splitting of nucleosomes before transcription
depending from actif DNA-strand
|
|
|
Transcription from minus-strand
|
|
|
Transcription from plus-strand
|
|
|
Orphans (A):
2 x H2B
2 x H3
1 x Nucleoplasmin
Placeholder (A):
2 x H2A
2 x H4
1 x Nucleoplasmin
|
|
|
Orphans (B):
2 x H2A
2 x H3
1 x Nucleoplasmin
Placeholder (B)
2 x H2B
2 x H4
1 x Nucleoplasmin
|
|
|
Nach dem in der Tabelle beschriebenen Muster teilen sich die Oktamere dergestalt, dass abhängig vom zu transkribierenden Strang - ein bestimmter Teil der Histone entweder am plus-Strang oder am minus-Strang verbleibt. Der Gegenstrang ist frei von Histonen und wird transkribiert. Die Position der an die DNA gebunden bleibenden Histone verändert sich dabei nicht. Durch die Reassemblierung nach dem Durchgang der RNAPII wird das alte spacing, die ursprüngliche Nukleosomenkonfiguration wieder hergestellt.
Die Funktion von Nukleoplasmin.
Nukleoplasmin ist bereits seit 1978 (Laskey et al.) als molekulares Chaperon bekannt, das eine wichtige Rolle bei der Assemblierung von Histonen zu Oktameren spielt. Iihara et al. konnten zeigen, dass eine der Aufgaben von Nukleoplasmin in der Verhinderung der vorzeitigen Aggregation von Histonen und DNA besteht.249
Dies dürfte die Funktion von Nukleoplasmin auch im hier beschriebenen Prozess des Nukleosomen-Splittings sein: seine Inkorporation in die Orphan- bzw. die Platzhalter-Komplexe sorgt für den Zusammenhalt der Komponenten, bis sie wieder an den ursprünglichen Platz zurückkehren können. Diese reversible Bindung der Orphan- respektive Platzhalter-Komponenten durch Nukleoplasmin ist wahrscheinlich abhängig von einer spezifischen Modifizierung von Nukleoplasmin.
Hox-Proteine und ihr Einfluss auf die Genexpression.
Wie ich an anderer Stelle ausgeführt habe, haben alle vielzelligen Lebenwesen sich aus einer einzigen Urzelle entwickelt. Dieser Entwicklung lagen Mechanismen zugrunde, die ich in dieser Arbeit versucht habe zu beschreiben: insbesondere das System der Nukleosomengrössen, das die Zusammenfassung von Genen zu Transkriptionseinheiten oder - gruppen und die koordinierte Steuerung ihrer Expression durch hormonelle Impulse erlaubt, ist ein Teil des gemeinsamen Bauplans, welcher die Entwicklung aller Lebewesen bestimmt.
Dass es trotz der zum Teil grossen Übereinstimmungen der Genome, die bei Schimpanse und Mensch zum Beispiel 98,7% beträgt, zur Entwicklung ganz verschiedener Arten kommen konnte, muss also andere Ursachen haben. Eine dieser Ursachen habe ich im Kapitel Der Histon-DNA-Code genannt: während der Grad der Nutzung (messbar an der Menge der produzierten mRNA) der Gene von Leberzellen bei Schimpanse und Mensch kaum differiert, ist er in Zellen des Gehirns eklatant: Die Gene in menschlichen Gehirnzellen werden in viel größerem Umfang (etwa um den Faktor 4 stärker) ausgenutzt durch exzessives alternatives Spleißen z.B. als die korrespondierenden Gene der Primaten.
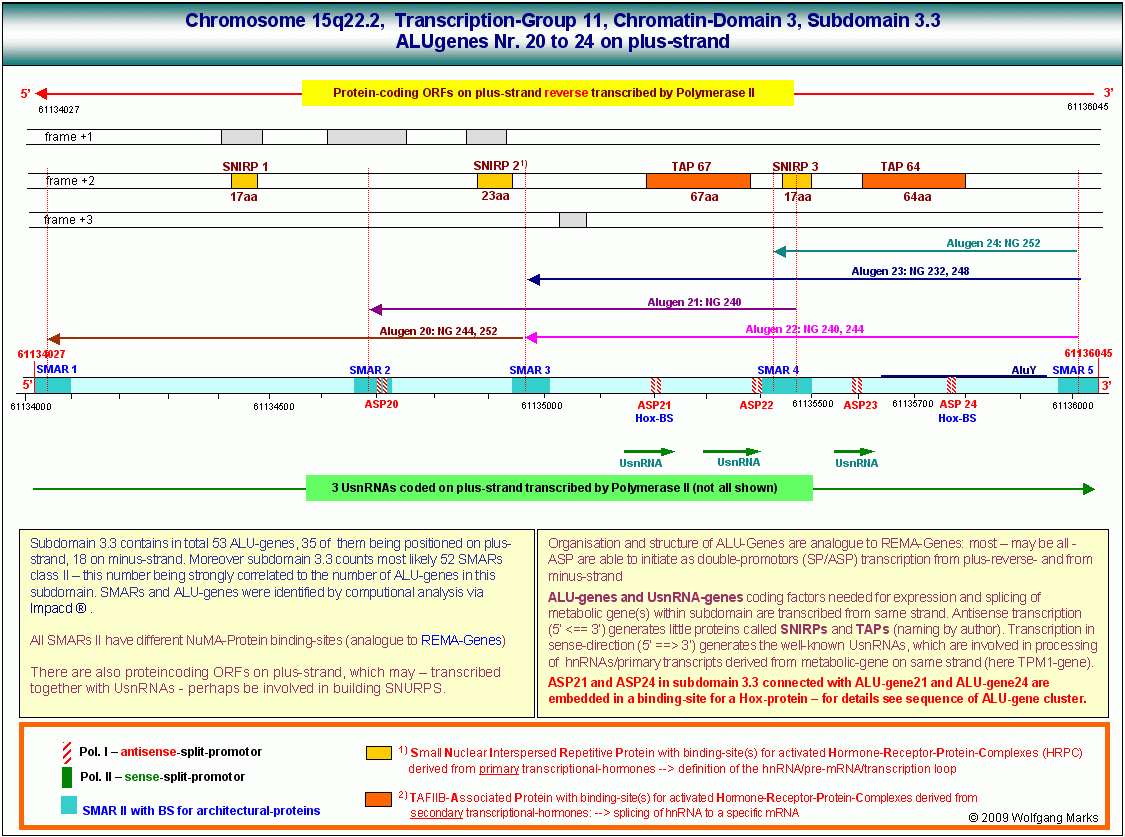
Aber auch durch die Evolution der Spleissmechanismen und die damit verbundene Evolution der ALU-Gene allein lässt sich die Diversifizierung der Arten nicht schlüssig erklären. Es muss ein System geben, das dem in dieser Arbeit skizzierten logisch-logistischen System der Nukleosomengrössen übergeordnet ist - es muss einen Informationslayer geben, der die Netzwerke von REMA-Genen und ALU-Genen und damit die Genexpression sowohl an der Basis (der Formatierung von Chromatin-Domänen durch REMA-Gen kodierte remodeling-Maschinen) als auch auf einer höheren Ebene (der Regulation der Transkription und des Spleissens durch ALU-Gene) so zu steuern vermag, dass bei gleichem Baumaterial die unterschiedlichsten Baupläne realisiert werden können.
Diesen übergeordneten Informationslayer stellt zweifellos das Hox-Gen-System dar. Obwohl es bis heute nicht gelungen ist, die Wirkungsweise dieses Systems, die Art seiner Einwirkung auf die morphologische Entwicklung der Lebewesen zu entschlüsseln, steht ausser Zweifel, dass dieses System den Masterplan für die Diversifizierung der Arten repräsentiert.
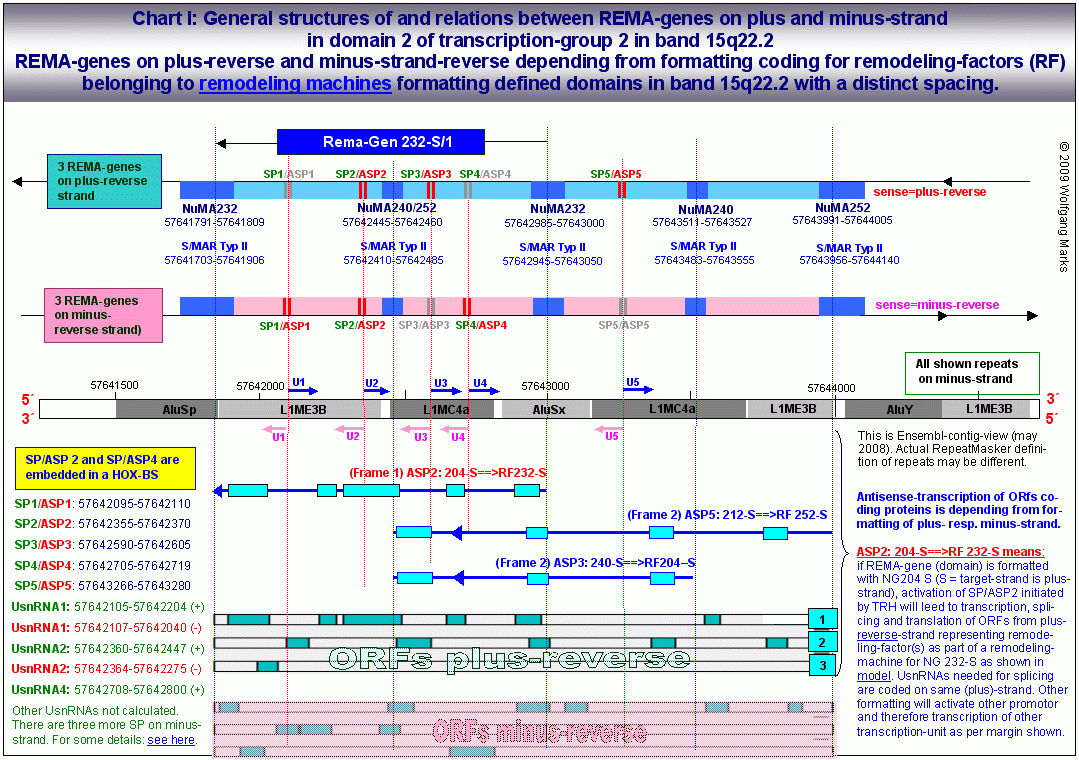
Wie auf den beiden obenstehenden Grafiken zu sehen, habe ich sowohl bei der Analyse der REMA_Gene als auch der ALU-Gene Homöoboxen, also Bindestellen für Hox-Proteine identifiziert, die mit den Positionen von sense- und antisense-Promotoren nahezu deckungsgleich sind.
Durch die Bindung von Hox-Proteinen an diese Bindestellen in REMA- beziehungsweise ALU-Genen kann die Zelle demnach Einfluss auf die beiden essentiellen Prozesse nehmen, welche die Genexpression genomweit determinieren:
Die Formatierung einer Chromatin-Domäne ist von REMA-Genen bestimmt, deren Promotoren in chromosomen-übergreifenden Netzwerken zusammengefasst sind.
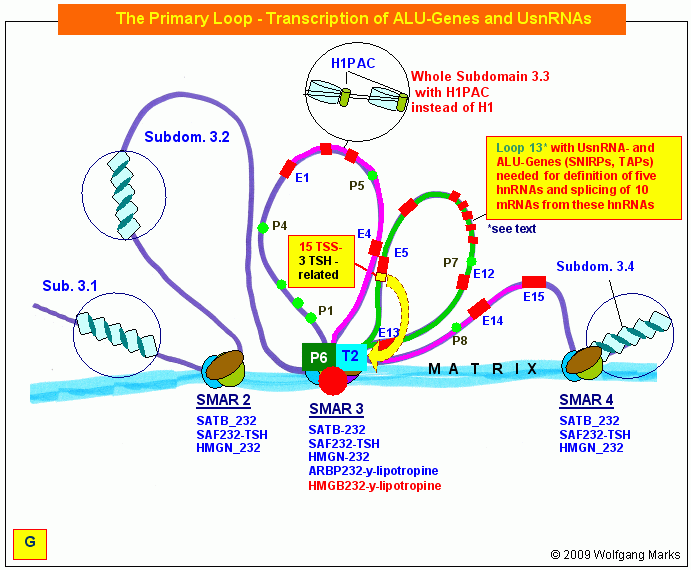
Die durch REMA-Gene kodierten Remodeling-Maschinen formatieren Chromatin-Domänen zell- und entwicklungsabhängig mit einer spezifischen, epigenetisch determinierten Nukleosomengrösse. Diese bestimmt die Auswahl von Promotor und Terminator und definiert die primäre Transkriptionsschleife, in der sowohl die UsnRNAs, als auch die ALU-Gene und damit die spezifischen Transkriptionsfaktoren (SNIRPs ud TAPs) kodiert sind, welche die Expression und das Processing des in der Transkriptionsschleife gelegenen metabolen Gens steuern.
Die Bindung von HOX-Proteinen an die Promotoren von REMA- und ALU-Genen stellt den einfachsten Weg dar, diese beiden für die Expression der metabolen Gene entscheidenden Gensysteme genomweit zu steuern und repräsentiert daher zweifellos den weiter oben postulierten globalen Informationslayer, der die morphologische Entwicklung aller Lebenwesen determiniert.
Ob durch die Bindung von Hox-Proteinen an Homöoboxen in REMA- und ALU-Genen die Expression der konnektierten REMA- bzw. ALU-Gene reprimiert oder aktiviert wird, ist für die Funktion und die Logik des Systems nur von untergeordneter Bedeutung und kann für den Augenblick dahingestellt bleiben.
Zusammenfassung .
Die Definition einer zellspezifischen mRNA ist also von einer Vielzahl von Faktoren bestimmt, von denen ich die meisten und vermutlich entscheidenden in dieser Arbeit charakterisiert und beschrieben habe.
Dazu gehören Architekturproteine, Hormon-Rezeptorprotein-Komplexe, nicht genauer bekannte Mediatoren oder Modulatoren und spezifische SNIRPs und TAPs, welche - bedingt durch ihre Verknüpfung mit primären bzw. sekundären Transkriptionshormonen - spezifische Funktionen einmal bei der Definition des Primärtranskripts, zum andern bei der Definition der zu spleissenden Exons haben. Aber auch die spezifischen UsnRNAs, die in der primären Transkriptionsschleife kodiert sind, haben wahrscheinlich Anteil an der Auswahl der Exons, die zu einer zellspezifischen mRNA gespleisst werden. Ihre Positionierung in der primären Transkriptionsschleife ist sicher nicht zufällig. Zufällig sind sicher auch nicht die arithmetischen Relationen zwischen Core-Promotor und TSS, ebenso wenig wie die zwischen TSS und DPE oder die zwischen poly-A-Signal/Terminator und poly-Adenylierungs-Stelle.
Auch die Länge des sogenannten poly-A-Schwanzes, die Zahl der an die mRNA angehefteten Adenyl-Reste dürfte beim processing des Primärtranskripts/hnRNA zu einer bestimmten mRNA eine Rolle spielen. Diese Relationen und ihren Einfluss auf die Auswahl der Exons und die Art ihrer Verarbeitung werde ich vielleicht zu einem späteren Zeitpunkt untersuchen.
Vom Gen zur messenger-RNA so habe ich das Thema dieser Arbeit untertitelt. Ich habe versucht, die Abläufe und Mechanismen zu beschreiben, die zu epigenetisch determinierten Chromatinstrukturen und zur zell- und entwicklungsspezifischen Transkription von Genen führen. Zwei vom Autor entdeckte Gensysteme die REMA- und die ALU-Gene - die daran entscheidenden Anteil haben, habe ich detailliert beschrieben und die Netzwerke charakterisiert, in denen sie zusammengefasst sind. Die Basis dieser Netzwerke stellt das System der Nukleosomengrössen und der Histon-DNA-Code dar, den ich bereits 1990 postuliert und beschrieben habe einige Jahre bevor Andere die gleiche Hypothese aufgestellt haben.
Das System der Nukleosomengrössen und seine Verknüpfung mit dem kaskadierten Hormonsystem und die Systematik, mit der die Zelle die horizontale und die vertikale Ebene der Genexpression logisch miteinander verbindet, waren weitere Schwerpunkte dieser Arbeit.
Über die Funktion der SMARs, der REMAKEs, ihre Klassifizierung, ihre Struktur und die Art ihrer Bindestellen für Architektur- und andere mit der Transkription korrelierte Proteine habe ich im Kapitel Die Struktur der DNA referiert. Auch über die Abläufe während eines differentiellen Zellzyklus, über Silencing und Imprinting, das Verpackungssystem der Zelle habe ich ausführlich berichtet. Und schliesslich habe ich am konkreten Beispiel der Transkription des TPM1Gens den Weg vom Chromomer zur Transkriptionsschleife und zur Transkription einer zellspezifischen messenger-RNA Schritt für Schritt beschrieben und die daran beteiligten Faktoren und Prozesse charakterisiert.
Die vorliegende Arbeit ermöglicht damit einen umfassenden, revolutionären Einblick in Aufbau und Struktur des menschlichen Genoms und einen neuen Zugang zum Verständnis des vielschichtigen Netzwerks Genom sowie der komplexen Mechanismen der differentiellen Genexpression.
|
|
|
243Die Hormonkette kann auch nur zweistufig sein...
244Jun Ma: Gene Expression and Regulation; Springer-Verlag in coop. with Higher Education Press (HEP), Beijing, China; 2006, ISBN: 978-0-387-33208-6
245Dieses Kapitel ist auch über die GOOGLE-Buchsuche einsehbar: http://books.google.de/books?id=XVFrJyfnRBwC&source=gbs_navlinks_s
246PC2 ist ein Komplex, der ursprünglich aus HeLa-Zellkernextrakten isoliert wurde. Er hat ein Molekulargewicht von rund 500 kDa. Die Funktion von PC2 als Koaktivator ist abhängig von der Anwesenheit anderer Kofaktoren, zum Beispiel von TAFs in Verbindung mit PC3 und PC4. Der PC2 gehört zu den sogenannten kleinen humanen Mediator-Komplexen.
247Robzyk K, Recht J, Osley MA. Rad6-dependent ubiquitination of histone H2B in yeast.Science. 2000 Jan 21, 287 (5452), 501504; PMID 10642555.
248Sun ZW, Allis CD. Ubiquitination of histone H2B regulates H3 methylation and gene silencing in yeast. Nature. 2002 Jul 4, 418 (6893), 104108; PMID 12077605.
249Effect of nucleoplasmin on a nucleosome structure; IIHARA Akiko; SATO Koji ; HOZUMI Kentaro; YAMADA Masanori; YAMAMOTO Hiroyuki ; NOMIZU Motoyoshi ; NISHI Norio; Polymer journal 2002, vol. 34, no3, pp. 184-193
|
|
|

{kind=link}