|
Der Weg zur zellspezifischen mRNA: Promotor und Terminator werden ein Paar.
Wer oder was ist ein Promotor?
Nach der Bindung des H1PACs liegt die Subdomäne 3.3 also als offene, transkriptionsfähige (aktivierte) 10nm-Nukleosomenkette mit dem spacing 232 vor, während die anderen drei Subdomänen nach wie vor aus einer kompakten, kondensierten Kette mit gebundenem H1 bestehen222. Anzahl und Art der Proteine, die über die SMARs 3 und 4 mit der Kernmatrix assoziiert sind, hat sich nicht verändert.
Bevor ich über den nächsten Schritt, die Bindung eines spezifischen Promotors und eines spezifischen Terminators an die Matrix, bzw. die SMAR3 referiere, sind einige Anmerkungen zum aktuellen Promotor-Diskurs erforderlich:
Im humanen Genom sind zur Zeit etwa 775 verschiedene Promotoren bekannt, die sich durch grosse Unterschiede in ihrer Struktur und in ihrer Sequenz auszeichnen - eine allgemeine eukaryotische Promotorsequenz ist deshalb nur schwer zu charakterisieren.
Eukariotische Promotoren sind offensichtlich sehr komplex aufgebaut - bis heute hat man mehrere unterschiedliche DNA-Motive identifiziert, die die Funktionalität eines Promotors determinieren sollen. Zu diesen Elementen gehören sogenannte Enhancer und Silencer, eine UAS (upstream activating sequence), das BRE (TFIIB-recognition Element), die TATA-Box und/oder das Initiator-Element (Inr) sowie ein downstream promotor element (DPE). Die upstream activating sequence kann aus einer CAAT-Box bestehen oder aus einer GC-Box auch Sp1-Box genannt.
Nicht alle Elemente finden sich in allen Promotoren, bei vielen fehlt die TATA-Box diese Promotoren weisen stattdessen ein Initiator-Element auf. Manche Promotoren verfügen allerdings sowohl über eine TATA-Box, als auch über ein Initiator-Element. Der sogenannte Core- oder Minimal-Promotor soll das BRE, die TATA-Box bzw. das Inr und das DPE umfassen und zur Initiation einer basalen Transkription in vitro ausreichend sein.
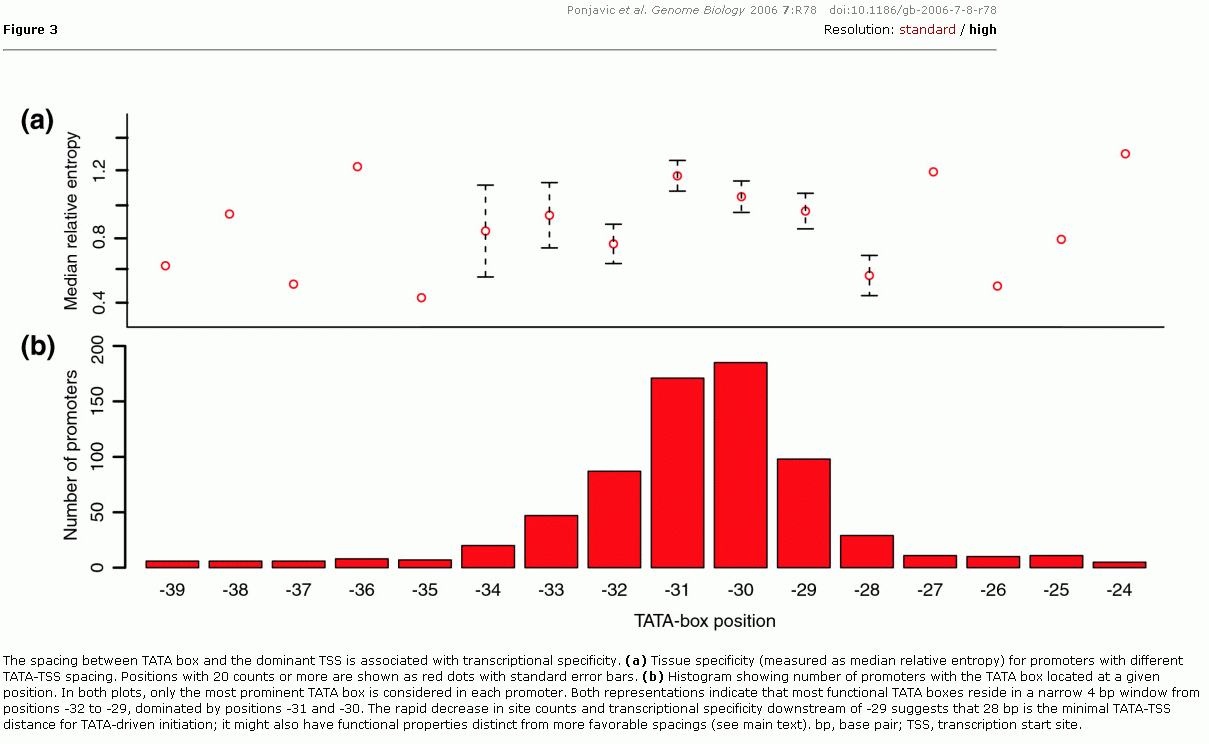
Während Enhancer- und Silencer-Elemente mehrere 1000bp stromaufwärts lokalisiert sein können und die UAS einige hundert bp stromauf, gehen fast alle Transkriptionsmodelle davon aus, dass der Transkriptionsstartpunkt, die TSS, in unmittelbarer Nähe der TATA-Box liegt. Zumeist wird der Abstand zwischen TATA-Box und TSS mit rund 30 bp angegeben. Ponjavic et al.223haben an Hand des Maus-Genoms für eine grosse Zahl von TATA-Boxen den mittleren Abstand zur TSS berechnet. Im abstract zu ihrer Arbeit schreiben sie:
Using a comprehensive set of 5.66 × 106 sequenced 5' cDNA ends from diverse tissues mapped to the mouse genome, we found that the TATA-TSS distance is correlated with the tissue specificity of the downstream transcript. To achieve tissue-specific regulation, the TATA box position relative to the TSS is constrained to 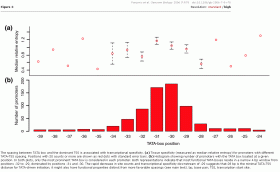 a narrow window (-32 to -29), where positions -31 and -30 are the optimal positions for achieving high tissue specificity. Slightly larger spacings can be accommodated only when there is no optimally spaced initiation signal; in contrast, the TATA box like motifs found downstream of position -28 are generally nonfunctional. The strength of the TATA binding protein-DNA interaction plays a subordinate role to spacing in terms of tissue specificity. Furthermore, promoters with different TATA-TSS spacings have distinct features in terms of consensus sequence around the initiation site and distribution of alternative TSSs. Unexpectedly, promoters that have two dominant, consecutive TSSs are TATA depleted and have a novel GGG initiation site consensus. a narrow window (-32 to -29), where positions -31 and -30 are the optimal positions for achieving high tissue specificity. Slightly larger spacings can be accommodated only when there is no optimally spaced initiation signal; in contrast, the TATA box like motifs found downstream of position -28 are generally nonfunctional. The strength of the TATA binding protein-DNA interaction plays a subordinate role to spacing in terms of tissue specificity. Furthermore, promoters with different TATA-TSS spacings have distinct features in terms of consensus sequence around the initiation site and distribution of alternative TSSs. Unexpectedly, promoters that have two dominant, consecutive TSSs are TATA depleted and have a novel GGG initiation site consensus.
Das heute geläufige Modell für die Aktivierung der Transkription geht von der Bildung eines sogenannten PICs (Pre-Initiation-Complex) aus, der sich aus generellen Transkriptionsfaktoren zusammensetzt. Dies soll am proximalen Promotor (auch Core-Promotor genannt) geschehen, der sich, wie bereits erwähnt, in einem Abstand von ca. 30 bp vor der TSS befinden soll. Das aktuelle Transkriptionsmodell stellt die Bindung des sogenannten TATA-Box-Bindeproteins TBP224 (das aber nicht nur an Promotoren mit TATA-Box-Motiv andockt) an den Core-Promotor an den Anfang, bei TATA-Box gesteuerten Genen wäre dieser Core-Promotor die TATA-Box selbst.
Allerdings gibt es seit einigen Jahren vermehrt Diskussionen über die Frage, wo die generellen Transkriptionsfaktoren (GTF) denn nun tatsächlich das erste Mal mit der DNA in Kontakt treten: Szutorisz et al. haben im November 2005 einen interessanten Übersichts-Artikel zu diesem Thema veröffentlicht225, in welchem sie eine Zusammenfassung eigener Arbeiten und Arbeiten anderer zu diesem Thema präsentieren, die das lange Zeit als unumstösslich geltende Paradigma, dass die generellen Transkriptionsfaktoren TFIIA, TFIIB, TFIID, TFIIE, TFIIF und TFIIH an einen Core-Promotor binden, der sich etwa 30 Basenpaare upstream der Transkriptions-Startstelle befindet, in Frage stellen und konterkarieren.
Seitdem die relevanten Untersuchungen nicht mehr mit in vitro-Systemen, sondern mit standardisierten Zellkultur- und Maus-Modellen durchgeführt werden, wird mehr und mehr evident, dass der proximale oder Core-Promotor nicht die alleinige Anlaufstelle für die GTFs, nicht der alleinige Ort sein kann, an dem sich das Transkriptosom bildet im Gegenteil: vor allem in sich differenzierenden Zellen während der Ontogenese sind Enhancer-Elemente/Upstream Activating Sequences wahrscheinlich die bevorzugten - wenn nicht die alleinigen - primären Bindestellen für (einen Teil der) GTFs.
Hätte ich die Arbeiten, die diese Beobachtungen untermauern226, bereits im Jahr 1999 oder 2000 gekannt, hätte ich mir wahrscheinlich viele schlaflose Nächte und viele Rechenläufe erspart - standen meine Promotor-Berechnungen (siehe Abbildung) doch in eklatantem Widerspruch zum Core-Promotor-Paradigma, das den PIC oder die Bindestelle für die GTFs in die unmittelbare Nachbarschaft der TSS plaziert227.
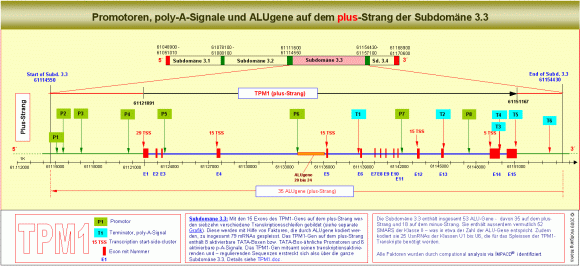 |
computational analysis der Subdomäne 3.3 mit IMPACD®
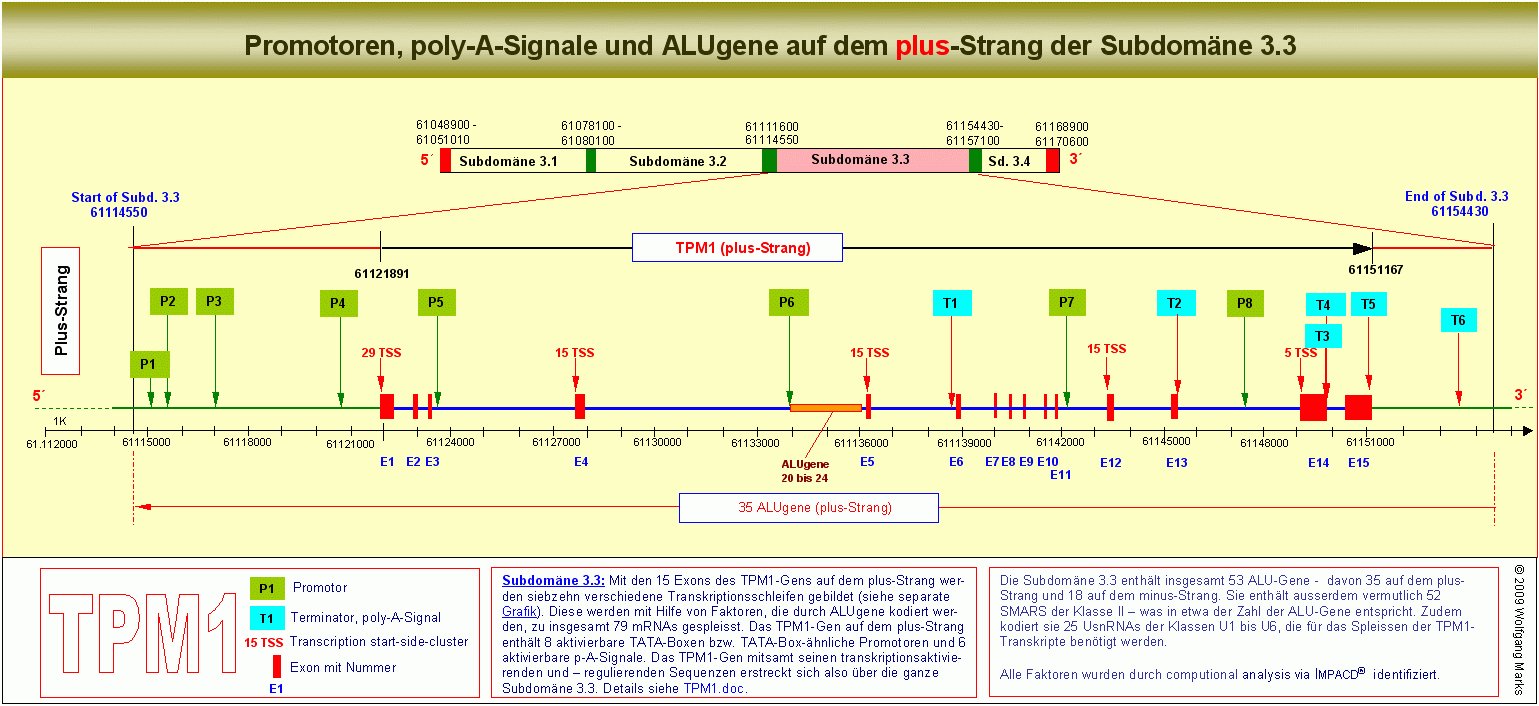
Die Subdomäne 3.3 - die eingerahmt wird von zwei SMARS der Klasse III enthält insgesamt 53 ALU-Gene - davon 35 auf dem plus-Strang und 18 auf dem minus-Strang, ausserdem 52 SMARS der Klasse II, welche ALU-Gene oder REMA-Gene begrenzen und Bindestellen für NuMA-Proteine enthalten, sowie 10 REMA-Gene, davon 5 auf dem plus- und 5 auf dem minus-Strang. Zudem sind 25 UsnRNAs in der Subdomäne kodiert, die für das Spleissen der TPM1-Transkripte benötigt werden.
Die Subdomäne 3.3 enthält ausserdem 8 aktivierbare TATA-Boxen bzw. TATA-Box-ähnliche Promotoren, 6 Terminatoren bzw. poly-A-Signale und insgesamt 79 Transkriptions-Startstellen (TSS), die in 5 Clustern vor und in den Exons 1, 4, 5, 12 und 14 zusammengefasst sind.
Wie auf der obigen Grafik und in der kommentierten TPM1-DNA-Sequenz235 zu sehen ist, sind die von mir identifizierten Promotoren und Terminatoren des TPM1-Gens über die ganze Subdomäne 3.3 verteilt. Der am weitesten stromauf liegende Promotor P1 befindet sich am Beginn der Subdomäne 3.3 etwa 7000 Basenpaare vor dem Exon 1. Der letzte Terminator, das am weitesten stromab liegende poly-A-Signal T6, befindet sich weit jenseits des Exons 15 kurz vor dem 3-Ende der Subdomäne. Es ist nicht zu übersehen: keiner der acht von mir mittels IMPACD® berechneten und definierten Promotoren befindet sich in unmittelbarer Nachbarschaft zu einer bekannten TSS oder zu einem der von mir berechneten TSS-Cluster. Der geringste Abstand zwischen einem Promotor (nach meiner Definition) und einer TSS des TPM1-Gens beträgt nach diesen meinen Berechnungen ca. 1000bp der grösste ca. 7000 Basenpaare.
Konnte ich mir den Widerspruch zwischen meinen Untersuchungsergebnissen und den in den Lehrbüchern festgeschrieben Paradigmen in den Jahren bis 2000 nicht erklären, löst sich dieser heute wortwörtlich in Wohlgefallen auf, weil sich zeigt, dass sowohl meine Berechnungen, als auch die Schlussfolgerungen daraus richtig waren. Enhancer-Regionen oder Upstream Activating Sequences stellen sich heute zumindest bei differentiell exprimierten (polycistronischen) Genen als die eigentlichen, die wahren Promotoren heraus (wenn man Promotoren als die primären Andockstellen für den PIC und die RNAPII definiert und nicht als den Startpunkt für die Transkription einer spezifischen mRNA).
Warum das so ist und auch gar nicht anders sein kann, werde ich in den folgenden Abschnitten versuchen zu zeigen.
Der Weg zur zellspezifischen mRNA: Gene looping.
Dass gene-looping kein seltenes Ereignis im Leben einer eukariontischen Zelle darstellt, geht aus zahlreichen neueren Veröffentlichungen hervor, von denen ich hier nur eine zitiere.228 Singh und Hampsey beschäftigen sich in ihrer Arbeit mit der Funktion des allgemeinen Transkriptionsfaktors TFIIB bei der Bildung einer Schleife zwischen Promotor und Terminator in S. cerevisiae. Sie kommen dabei zum Ergebnis, dass die Bildung einer Schleife zwischen Promotor229 und Terminator zwingend von der Bindung des TFIIB an den Terminator abhängt. Erst diese Bindung führt zu einem crosslinking zwischen Terminator und Promotor, also zur Bildung der Schleife. Die Autoren kommen weiter zu dem Schluss, das gene looping kein Phänomen sein kann, das auf ein bestimmtes Gen beschränkt ist, sondern ein normaler Vorgang innerhalb des Transkriptionsgeschehens sein muss. Gene looping ist nach Singh und Hampsey auch nicht auf besonders lange Gene konzentriert, sondern tritt zum Beispiel auch beim Gen HEM3 auf, das nur 1,0 kb aufweist.
Singh und Hampsey konnten auch nachweisen, dass gene looping nicht die Folge von Interaktionen zwischen DNA-Sequenzen innerhalb des Gens ist, sondern einzig und allein eine Folge des Zusammenbringens von Terminator und Promotor. Sie schliessen die Diskussion ihrer Arbeit mit der These, dass gene looping wahrscheinlich ein generelles Merkmal von Genen ist, die durch die RNAPII transkribiert werden.
Promotor und Terminator: eine fesselnde Beziehung.
Aus der in obigen Abbildung gezeigten Struktur des TPM1-Gens ergibt sich, dass es für die Kombination von Promotor und Terminator rein rechnerisch 48 mögliche Konstellationen gibt (8 x 6 = 48). Die Zahl von tatsächlich möglichen Kombinationen allerdings ist wesentlich geringer. Das hat einen einfachen Grund:
Dass die Promotoren und Terminatoren eines Gens nicht zufällig über die DNA verstreut sind, sondern über einen arithmetischen logos miteinander verknüpft sind, habe ich schon in Teil I und II postuliert (und werde es weiter unten belegen). Promotor und Terminator sind deshalb auch nicht beliebig kombinierbar, sondern bilden in Abhängigkeit von der Formatierung der Domäne logische Paare, die distinkte Transkriptionsschleifen oder -fenster definieren.
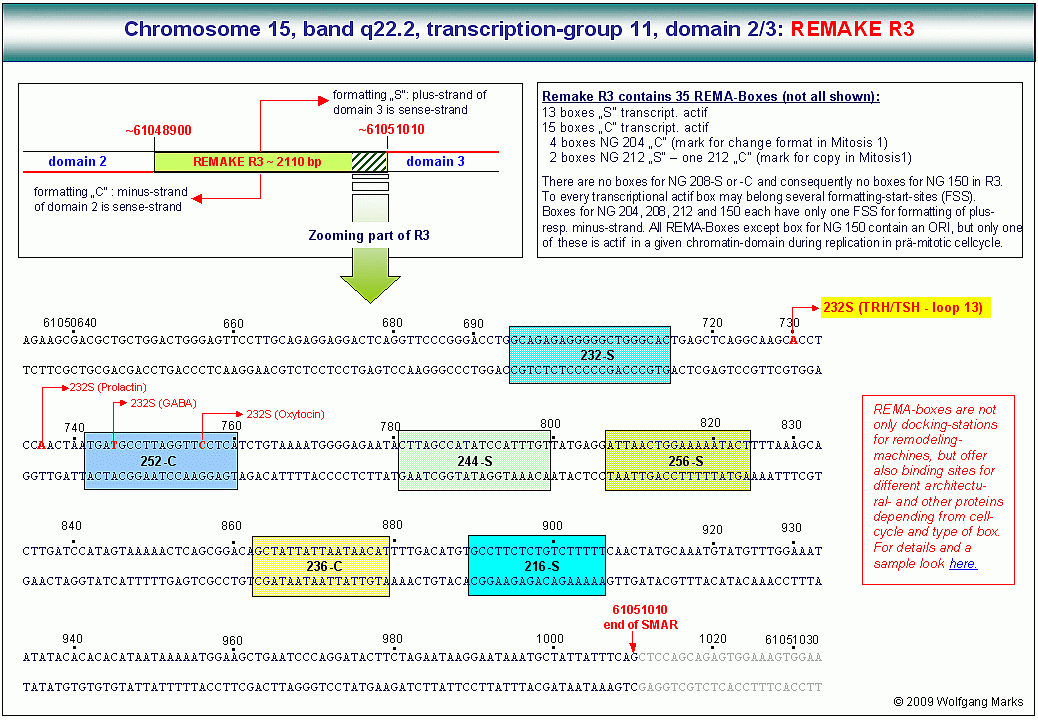
Ein Beispiel: wenn die Domäne 3 der TG 11 des Chromosomenbandes 15q22.2 von einem bestimmten Startpunkt aus im REMAKE dieser Domäne mit einer distinkten Nukleosomengrösse formatiert ist, wird ein bestimmtes Promotor/Terminator-Paar auf der linker DNA zwischen zwei Nukleosomen-Cores liegen, wo es für die Transkriptionsfaktoren erreichbar ist.
Natürlich wird es - formatierungsabhängig - Kombinationen zwischen einem bestimmten Promotor und zwei verschiedenen Terminatoren/poly-A-Signalen und vice versa geben. Die Formatierung allein schafft also keine eindeutige (Informatiker sagen: keine 1:1) Beziehung zwischen Promotor und poly-A-Signal. An der Installation einer 1:1 Beziehung zwischen Promotor und Terminator, an der Definition einer transkriptionsaktiven primären Transkriptionsschleife müssen also noch ein oder mehrere andere Faktoren beteiligt sein.
Einer dieser Faktoren ist nach meinen Analysen ein Architekturprotein der HMBG-Familie, im konkreten Fall das HMGB232-y-Lipotropin. Es wird über die Hormon-Triade TRH/TSH/y-Lipotropin aktiviert und bindet an den Terminator T2.
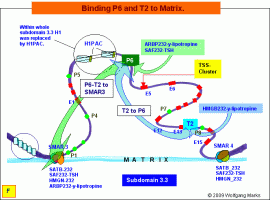 Die Bindung von HMGB232-y-Lipotropin an T2 führt T2 an den Promotor P6 heran, als Folge dessen bildet T2 mit P6 einen Komplex, der nach der Rekrutierung weiterer Architekturproteine (ARBP232-y-Lipotropin und SAF232-TSH) schliesslich an der SMAR3/Kernmatrix fixiert wird. An den SMAR3/Matrix-Komplex binden nun die GTFs, eine Topo-Isomerase, eine Helikase und die RNAP II. Durch die Fixierung des T2/P6-Aggregates an die Matrix entstehen demnach zwei Schleifen, von denen die eine den inaktiven Teil der Subdomäne 3.3 vom 5-Ende bis zum Promotor P6 umfasst, die andere den aktiven Teil der Subdomäne zwischen P6 und T2. Diesen Teil zwischen P6 und T2 nenne ich Die Bindung von HMGB232-y-Lipotropin an T2 führt T2 an den Promotor P6 heran, als Folge dessen bildet T2 mit P6 einen Komplex, der nach der Rekrutierung weiterer Architekturproteine (ARBP232-y-Lipotropin und SAF232-TSH) schliesslich an der SMAR3/Kernmatrix fixiert wird. An den SMAR3/Matrix-Komplex binden nun die GTFs, eine Topo-Isomerase, eine Helikase und die RNAP II. Durch die Fixierung des T2/P6-Aggregates an die Matrix entstehen demnach zwei Schleifen, von denen die eine den inaktiven Teil der Subdomäne 3.3 vom 5-Ende bis zum Promotor P6 umfasst, die andere den aktiven Teil der Subdomäne zwischen P6 und T2. Diesen Teil zwischen P6 und T2 nenne ich 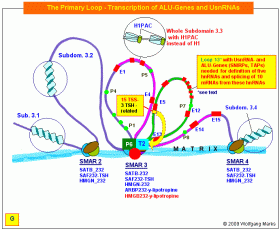 die primäre Transkriptionsschleife in der Abbildung G ist er als loop 13 bezeichnet. Mit dieser loop 13 werde ich mich nun intensiver beschäftigen. die primäre Transkriptionsschleife in der Abbildung G ist er als loop 13 bezeichnet. Mit dieser loop 13 werde ich mich nun intensiver beschäftigen.
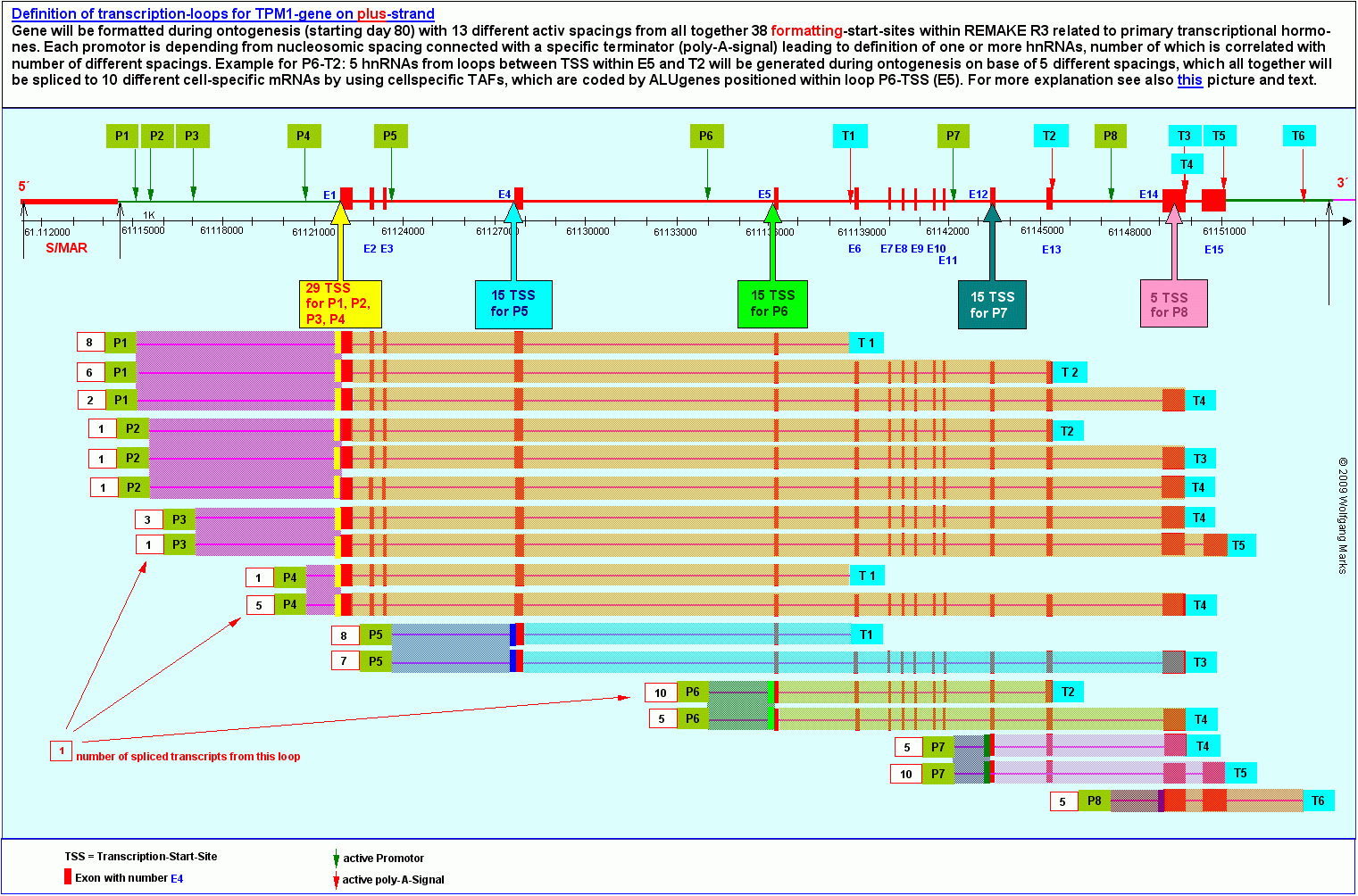
Loop 13 ist eine von 17 primären Transkriptionsschleifen, die ich mittels IMPACD® für das TPM1-Gen berechnet habe. Diese 17 Transkriptionsschleifen oder fenster repräsentieren nach meinen Berechnungen insgesamt 38 hnRNAs230 oder Primärtranskripte, die mit 13 verschiedenen REMA-Boxen (das entspricht 13 verschiedenen Nukleosomengrössen und Formatierungshormonen) und 38 verschiedenen Formatierungs-Startstellen im REMAKE R3 am 5-Ende der Domäne 3 verknüpft sind. Sie werden mit Hilfe von UsnRNAs und ALU-Gen kodierten SNIRPs und TAPs von 79 verschiedenen TSS (transcription start sites) aus zu 79 verschiedenen messenger-RNAs gespleisst.
Das bedeutet, dass das TPM1-Gen während der Ontogenese wahrscheinlich auch in insgesamt 79 verschiedenen Zellen exprimiert wird.231
Diese Zahlen erscheinen zunächst hoch allerdings zählen der UCSC Genome Browser und ENSEMBLE für TPM1 auch über 60 potentielle mRNAs. Zudem wird das TPM1-Gen in vielen verschiedenen Zellen und fast während der gesamten Ontogenese exprimiert.
Die folgende Grafik zeigt die von mir berechneten Beziehungen zwischen Promotoren und Terminatoren des TPM1-Gens auf der Basis der von mir definierten Nukleosomengrössen (inkomplett, wird noch vervollständigt).
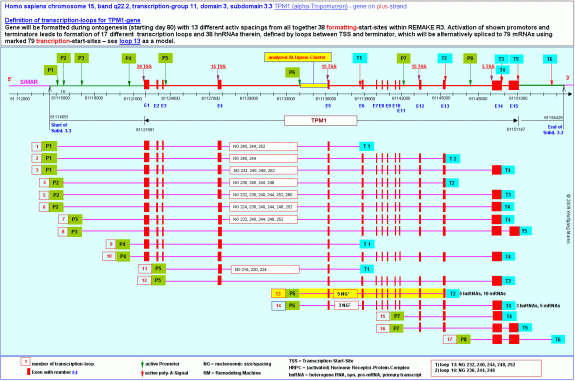 |
Die Expression des TPM1-Gens muss mit der Transkriptionsschleife 11 beginnen, die mit den Nukleosomengrössen 216, 220 und 224 korreliert ist. Damit fällt die Expression dieses Gens mit dem Anfang der Thymusperiode zusammen sie beginnt also kurz nach dem 80. Tag der Ontogenese. Die Bildung der Transkriptionsschleife 13 ist unter anderem mit dem Formatierungshormon TRH und dem primären Transkriptionshormon TSH verknüpft. Diese TRH/TSH-korrelierte hnRNA wird etwa ab dem 110. Tag der Ontogenese gebildet.
Die Formatierung definiert die primäre Transkriptionsschleife.
Die Transkriptionsschleife 13 die der Gegenstand dieser Untersuchung ist - kodiert nach meinen Berechnungen 5 Primärtranskripte232, aus denen in unterschiedlichen Zellen und zu unterschiedlichen Zeiten zehn verschiedene messenger-RNAs gespleisst werden. Diese 5 Primärtranskripte sind von der Formatierung der Domäne 3 mit einer der Nukleosomengrössen 232, 240, 244, 248 oder 252 abhängig. Das durch die Nukleosomengrösse 232 und die Hormone TRH/TSH definierte Primärtranskript wird etwa zwischen dem 110. Tag und dem 145. Tag, die daraus gespleisste y-Lipotropin-mRNA etwa zwischen dem 110. Tag und dem 120. Tag der Ontogenese gebildet.
Sehen wir uns nun die gelb markierte Transkriptionsschleife 13 etwas genauer an: sie reicht vom Promotor P6 bis zum poly-A-Signal bei T2 und umfasst die Exons 5 bis 13 und im Exon 5 einen TSS-Cluster aus 15 mittels IMPACD® berechneter Transkriptions-Startstellen. In dem ebenfalls gelb markierten Bereich vor dem Exon 5 befindet sich der von mir zuvor analysierte ALU-Gen-Cluster mit dem ALU-Gen 23, welches für das SNIRP3 und das TAP64 kodiert, die ich weiter oben eingehend untersucht und beschrieben habe. Zur Erinnerung: Das SNIRP 3 mit 17aa weist unter anderen eine Bindestelle für einen TSH-HRPK auf, das TAP64 unter anderen eine solche für das TSH-abhängige Hormon y-Lipotropin.
Dass der ALU-Gen-Cluster vor Exon 5 sich nicht zufällig in dieser genomischen Position und damit innerhalb der primären Transkriptionsschleife 13 befindet, liegt auf der Hand: der Schluss liegt nahe, dass die Proteine, die in diesem ALU-GenCluster kodiert sind, mit der Transkription und dem Spleissen der Exons 5 bis 13 zu verschiedenen messenger-RNAs verknüpft sind. Dies umso mehr, als die Transkriptionsschleife 13 durch die gleichen fünf Nukleosomengrössen (NG 232, 240, 244, 248, 252) definiert wird, welche auch mit der Aktivierung der ALU-Gen-Promotoren im markierten ALU-Gen-Cluster vor Exon 5 verknüpft sind.
Promotor-Gruppen und ihre Beziehung zu TSS-Clustern und ALU-Genen.
Wer die obige Abbildung genauer betrachtet hat, dem wird aufgefallen sein, dass die Promotoren 1 bis 8 sich in Gruppen einteilen lassen, die in Relation zu einem bestimmten Exon und zu einem Cluster von TSS stehen. So sind die Promotoren P1 bis P4 zum Beispiel mit den 29 Transkriptions-Startstellen stromauf von Exon 1 bzw. im Exon 1 verknüpft - P5 mit den 15 Startstellen vor/in Exon 4. Die gleichen Beziehungen gelten auch für die anderen 3 Promotor-Gruppen und die mit ihnen korrelierten TSS-Cluster.
Im TPM1-Gen gibt es nach meinen Berechnungen fünf solcher TSS-Cluster, die wie bereits gesagt - insgesamt 79 Transkriptions-Startstellen repräsentieren. Diese befinden sich vor und zum Teil in den Exons 1, 4, 5, 12 und 14. Im einzelnen:
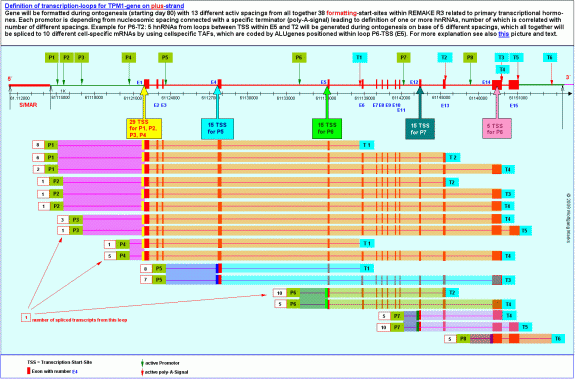 29 TSS vor/in Exon 1 29 TSS vor/in Exon 1
15 TSS vor/in Exon 4
15 TSS vor/in Exon 5
15 TSS vor/in Exon 12
5 TSS vor/in Exon 14
Die meisten dieser TSS sind unbekannt: lediglich vor und am Beginn von Exon 1 hat die European Promotor Database (EPD) einen Cluster von TSS definiert, in ENSEMBLE ist eine einzelne TSS vor Exon 4 vermerkt, die TSS-Cluster vor Exon 5, 12 und 14 sind meines Wissens bisher nicht bekannt.
Nach meinen Berechnungen kann ein Promotor der Gruppe 1 (P1, P2, P3, P4) nicht mit einer TSS zum Beispiel der Gruppe 2 oder der einer anderen Promotor-Gruppe verknüpft sein. Jede primäre Transkriptionschleife hat also a priori eine homogene, ununterbrochene lineare Struktur Spleissen und Verarbeitung einer hnRNA zu einer mRNA kann deshalb auch nur innerhalb der durch Promotor und Terminator gesetzten Grenzen stattfinden. Innerhalb dieser Grenzen, in der primären Transkriptionsschleife also, werden sowohl die Faktoren kodiert, die für die Definition einer bestimmten hnRNA notwendig sind (SNIRPs), als auch jene Faktoren, die für die Festlegung der TSS (TAPs) und das Spleissen des derart definierten Transkripts zu einer mRNA erforderlich sind (UsnRNAs).
Ein Beispiel: die ALU-Gen-Faktoren (SNIRPs, TAPs, UsnRNAs), die für die Definition, Transkription und Verarbeitung der Schleife 13 und 14 benötigt werden, können nicht upstream von Promotor P6 liegen, sondern nur innerhalb der Grenzen, die durch den Promotor P6 und den Terminator T2 (loop 13) bzw. T4 (loop 14) gebildet werden. Die Auswahl der zur Verfügung stehenden Transkriptionsfaktoren ist also ebenso wie die Auswahl der Exons durch die Organisation der Transkriptionschleife (des Transkriptionsfensters, würde mein Freund Ferenc Müller sagen), eingeschränkt und begrenzt. Die Nukleosomengrösse definiert somit letztlich auch die Auswahl der zellspezifischen TAFs, die zusammen mit Architekturproteinen und allgemeinen Transkriptionsfaktoren an der Bildung des Transkriptosoms und der Generierung einer zellspezifischen messenger-RNA beteiligt sind.
Die arithmetischen Beziehungen zwischen Promotor,
Terminator und TSS.
Die phasengetreue Formatierung definierter Abschnitte der DNA mit distinkten Nukleosomengrössen und die Koppelung dieses Systems mit dem kaskadierten, hierarchischen Hormonsystem des menschlichen Körpers ist die Basis der differentiellen Genexpression. Durch eine definierte Formatierungsstartstelle (FSS) im REMAKE einer Chromatin-Domäne werden die Positionen der Oktamere auf der DNA und damit die Regionen festgelegt, die in dieser Domäne für Chromatin-Modulatoren, Transkriptionsfaktoren und Regulatorproteine zugänglich sein sollen.
Wie ich mehrfach ausgeführt habe, ist die Expression eines Gens vor allem davon abhängig, dass die hotspots des betreffenden Gens dergestalt auf der linker-DNA plaziert sind, dass sie von aktivierenden Faktoren angesprochen werden können. Ich habe das an anderer Stelle den inhärenten genomischen logos genannt und behauptet, dass es eine arithmetische Beziehung zwischen allen DNA-Motiven geben muss, welche die Generierung einer spezifischen mRNA determinieren. Diese DNA-Motive schliessen die Formatierungs-Startstelle ebenso ein wie eine bestimmte TSS, einen Promotor, einen Terminator und andere Motive wie ALU- und REMA-Gen-Promotoren oder Sequenzen, welche die Expression eines spezifischen Gens modulieren, hemmen oder verstärken.
Es ist nun an der Zeit, für diese Behauptung Belege zu liefern:
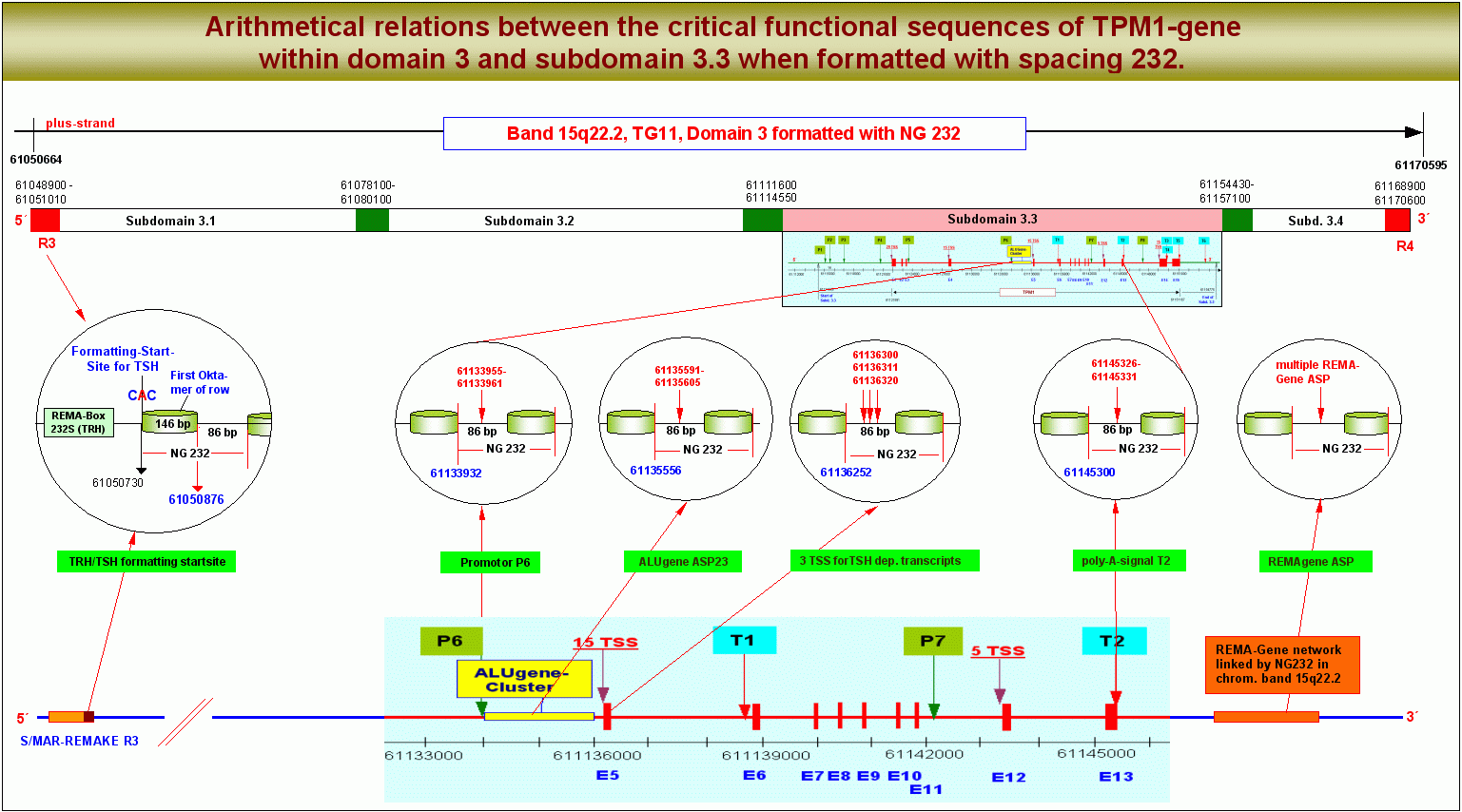
Dass die Relation zwischen der FSS im REMAKE einer Domäne und den verschiedenen hotspots der Genexpression in dieser Domäne tatsächlich auf einer arithmetischen Beziehung zwischen diesen Sequenzen beruht, zeigt die folgende Grafik. Sie bietet einen globalen Blick auf die Chromatin-Domäne 3 und setzt die Koordinaten von FSS, TSS, dem Promotor P6, dem poly-A-Signal T2 und dem ALU-Gen-Promotor ASP23 in Beziehung zur Nukleosomengrösse 232. Die angegebenen Zahlen beziehen sich auf die chromosomalen Koordinaten nach ENSEMBLE, sie sind mit denen bei NCBI identisch und lassen sich mit jedem Taschenrechner nachprüfen.
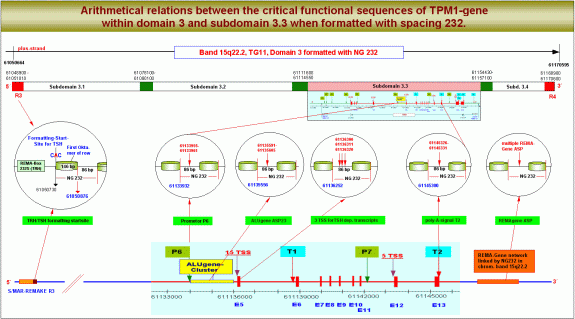 |
Arithmetische Korrelation zwischen den hotspots eines Gens, hier des TPM1-Gens. Wenn die Domäne 3 der Transkriptionsgruppe 11 des Chromosomenbandes 15q22.2 mit der Nukleosomengrösse 232 vom TSH-Startpunkt im REMAKE R3 aus formatiert wird, liegen der Promotor P6, der ALU-Gen ASP23, drei TSS, definiert durch drei von TSH abhängige sekundäre Transkriptionshormone und das poly-A-Signal T2 jeweils auf der linker-DNA zwischen zwei Nukleosomen-Cores. Ein Cluster von REMA-Genen im Chromosomenband 15q22.2 ist über die Nukleosomengrösse 232 zu einer Transkriptionseinheit zusammengefasst, die für eine epigenetisch definierte spezifische remodeling-Maschine kodiert.
Wenn die Domäne 3 mit der Nukleosomengrösse 232 vom TSH-Startpunkt aus formatiert wird, liegen der Promotor P6, der ALU-Gen ASP23, drei TSS, verknüpft mit drei von TSH abhängigen sekundären Transkriptions- oder Processinghormonen und das poly-A-Signal T2 demnach jeweils auf der linker-DNA zwischen zwei Nukleosomen-Cores. Auch ein oder mehrere REMA-Gene sind mit anderen REMA-Genen im Chromosomenband 15q22.2 über die Nukleosomengrösse 232 zu einer Transkriptionseinheit zusammengefasst, die für eine epigenetisch definierte spezifische remodeling-Maschine kodiert.
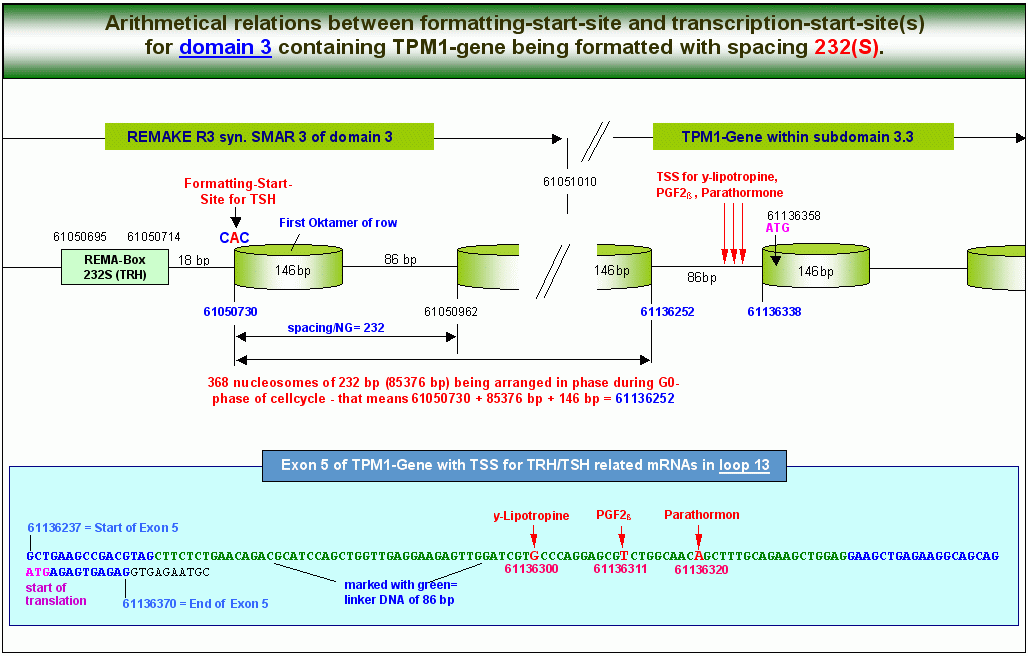
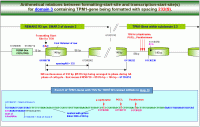 Die in der obenstehenden Grafik für die gesamte Domäne 3 demonstrierten Relationen zwischen den Hotspots des TPM1-Gens zeigt die nebenstehende Grafik für den TSH-Formatierungs-Startpunkt im REMAKE R3 und die mit diesem arithmetisch verknüpften drei TSS vor dem Startpunkt der Translation im Exon 5 noch einmal im Detail. Die in der obenstehenden Grafik für die gesamte Domäne 3 demonstrierten Relationen zwischen den Hotspots des TPM1-Gens zeigt die nebenstehende Grafik für den TSH-Formatierungs-Startpunkt im REMAKE R3 und die mit diesem arithmetisch verknüpften drei TSS vor dem Startpunkt der Translation im Exon 5 noch einmal im Detail.
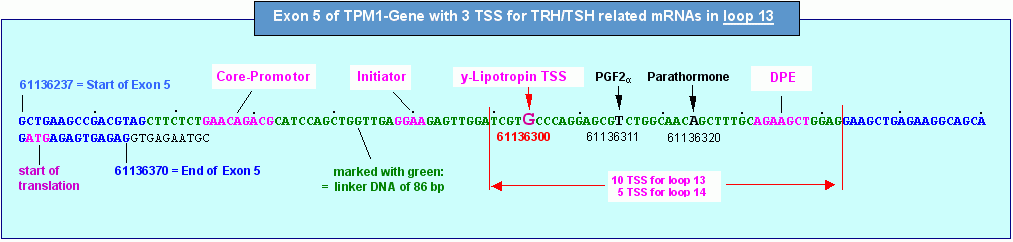
Die Abbildung unten schliesslich zeigt die genomische Struktur des plus-Strangs des Exons 5 mit den von mir berechneten DNA-Motiven, die in die Expression einer y-Lipotropin-mRNA involviert sind. Wenn das TPM1-Gen (die Domäne 3) vom TSH-Startpunkt aus im REMAKE R3 mit der NG 232 formatiert ist, befinden sich die grün markierten Nukleotide/Basenpaare wie in der Grafik zu sehen -auf der linker DNA zwischen zwei Nukleosomen-Cores. In diesem Bereich, auf der linker-DNA zwischen zwei Cores, befinden sich alle für die Initiation der Transkription relevanten DNA-Motive, also der sogenannte Core-Promotor, das Initiator-Element und die TSS für y-Lipotropin.
 |
Es ist deutlich zu erkennen, dass nicht nur die TSS, sondern auch der Core-Promotor und das Inititator-Element für die y-Lipotropin-TSS bei dieser Nukleosomenkonfiguration derart auf der linker-DNA plaziert sind, dass Transkriptionsfaktoren an diese Elemente binden können. Dies gilt auch für die beiden anderen von TSH abhängigen TSS. Da der Abstand zwischen Core-Promotor und TSS nach Ponjavic et al. aber auch nach meinen Berechnungen - zwischen 29 und 32 Basenpaaren liegt, verschieben sich Core-Promotor und Inr-Element zwar (auf die PGF2a- und die Parathormon-TSS bezogen) in Richtung 3-Ende des Exons, befinden sich aber auch dann noch innerhalb des Aktivierungsfensters, das bei Formatierung des TPM1-Gens mit der NG 232 durch die grün markierten 86 Basenpaare gebildet wird. Eindringlicher kann die Abhängigkeit der Transkriptionsaktivierung von der Formatierung eines Gens kaum demonstriert werden.
Auf das Transkriptionsfenster 13 (loop 13 zwischen P6 und T2) bezogen befinden sich weitere 7 TSS im gekennzeichneten Bereich vor dem Startcodon ATG. Diese TSS sind aber nicht mit dem TSH-Startpunkt im REMAKE R3 korreliert, sondern mit Startstellen, die mit einer von 232 verschiedenen233 Nukleosomengrösse und damit auch mit anderen Formatierungshormonen sowie anderen primären Transkriptionshormonen korreliert sind. Es versteht sich von selbst, dass sich bei einer Verschiebung der FSS auch das Aktivierungsfenster verschiebt es ist aber in allen Fällen so positioniert, dass die Gruppe, gebildet aus Core-Promotor, Initiator-Element und TSS innerhalb des Aktivierungsfensters liegt.
Aus diesen Ausführungen ergibt sich zwingend, dass bei Nukleosomengrössen, wie sie in der ersten Phase der Ontogenese der frühen Embryogenese gegeben sind, andere Mechanismen der Transkriptionsaktivierung zum Zuge kommen müssen. Bei der Nukleosomengrösse 176 zum Beispiel das entspricht einer Länge der linker-DNA von 30 Basenpaaren - ist das Aktivierungsfenster unter Berücksichtigung der Mindestabstände zu den Oktameren so klein, dass sich Core-Promotor-Motiv, Inr und TSS darin kaum unterbringen lassen234. Eine Lösung dieses Problems könnte darin liegen, dass die Transkriptionsaktivierung sich in dieser Phase der Zellentwicklung auf das Ansprechen eines Initiator-Elements (dann gleich Core-Promotor) und der TSS beschränkt.
Wenn man, was ich ja schon des öfteren getan habe, das menschliche Genom mit einer Datenbank vergleicht, dann könnte man sagen, dass durch die Formatierung einer Chromatin-Domäne mit einer definierten Nukleosomengrösse eine Variable definiert wird, welche die Ausführung des Teil-Programms Genaktivierung für diese Domäne, ihre Subdomänen und die in ihr gelegenen Gene determiniert.
Durch die Nukleosomengrösse und die damit verbundene An- oder Auswahl eines bestimmten Promotors und eines bestimmten poly-A-Signals wird also zum einen definiert, welche Exons und Introns in das zellspezifische Transkript eingehen können und welche nicht. Zum andern wird durch die Nukleosomengrösse aber auch definiert, welche Teile der genetischen Funktionseinheit zu einem bestimmten Zeitpunkt in einer gegebenen Zelle aktiviert und/oder transkribiert werden können. Diese genetische Funktionseinheit umfasst nicht nur Promotoren, poly-A-Signale, Exons und Introns oder ALU-Gene in der der jeweiligen Subdomäne, sondern auch die REMA-Gene des betreffenden Chromosomenbandes und gegebenenfalls auch ausserhalb dieses Bandes oder auf einem anderen Chromosom liegende Sequenzen, welche die Expression eines spezifischen Gens modulieren, hemmen oder verstärken können. Auch die Gene für die weiter oben beschriebenen Architekturproteine, die an der Aktivierung einer Transkriptionsgruppe, einer Domäne und oder Subdomäne beteiligt sind, zähle ich zu dieser Funktionseinheit.
Die Aktivierung aller genannten Faktoren muss zwingend an die Formatierung ihrer Chromatin-Domäne mit der gleichen Nukleosomengrösse gekoppelt sein wie die der Gene, an deren Aktivierung oder Regulation sie beteiligt sind.
Die Abbildung Arithmetical relations.. macht unmissverständlich deutlich, dass es zwischen den funktionell entscheidenden Sequenzen eines Gens eine auf distinkten Nukleosomengrössen basierende arithmetische Beziehung gibt, die sicherstellt, dass die transkriptionsaktivierenden, die transkriptionsterminierenden Sequenzen und die Sequenzen, die über die Aktivierung zellspezifischer Transkriptionsfaktoren entscheiden (ASP von ALU-Genen) nicht an ein Histon-Oktamer gebunden, sondern auf der linker-DNA frei zugänglich sind.
Der inhärente LOGOS, die arithmetische Ordnung, die sich von einem bestimmten Zeitpunkt an in definierten Nukleosomengrössen ausdrückte, war also von Anfang an das beherrschende Prinzip der genetischen Evolution der Eukarionten: nur weil alle funktionellen Sequenzen eines Gens soweit ihre Funktion von einer bestimmten Position innerhalb eines Nukleosoms abhängig ist - über definierte Nukleosomengrössen miteinander verbunden sind, führt ein Wechsel der Nukleosomengrösse auch zu einer veränderten Genexpression.
|

{kind=link}